 |
Code of Federal Regulations (Last Updated: November 8, 2024) |
 |
Title 40 - Protection of Environment |
|
Chapter I - Environmental Protection Agency |
|
SubChapter C - Air Programs |
|
Part 63 - National Emission Standards for Hazardous Air Pollutants for Source Categories |
|
Subpart UUU - National Emission Standards for Hazardous Air Pollutants for Petroleum Refineries: Catalytic Cracking Units, Catalytic Reforming Units, and Sulfur Recovery Units |
Appendix A to Subpart UUU of Part 63 - Determination of Metal Concentration on Catalyst Particles (Instrumental Analyzer Procedure)
-
Appendix A to Subpart UUU of Part 63 - Determination of Metal Concentration on Catalyst Particles (Instrumental Analyzer Procedure)
1.0 Scope and Application.
1.1 Analytes. The analytes for which this method is applicable include any elements with an atomic number between 11 (sodium) and 92 (uranium), inclusive. Specific analytes for which this method was developed include:
Analyte CAS No. Minimum detectable limit Nickel compounds 7440-02-0 <2 % of span. Total chlorides 16887-00-6 <2 % of span. 1.2 Applicability. This method is applicable to the determination of analyte concentrations on catalyst particles. This method is applicable for catalyst particles obtained from the fluid catalytic cracking unit (FCCU) regenerator (i.e., equilibrium catalyst), from air pollution control systems operated for the FCCU catalyst regenerator vent (FCCU fines), from catalytic reforming units (CRU), and other processes as specified within an applicable regulation. This method is applicable only when specified within the regulation.
1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from the analytical method.
2.0 Summary of Method.
2.1 A representative sample of catalyst particles is collected, prepared, and analyzed for analyte concentration using either energy or wavelength dispersive X-ray fluorescent (XRF) spectrometry instrumental analyzers. In both types of XRF spectrometers, the instrument irradiates the sample with high energy (primary) x-rays and the elements in the sample absorb the x-rays and then re-emit secondary (fluorescent) x-rays of characteristic wavelengths for each element present. In energy dispersive XRF spectrometers, all secondary x-rays (of all wavelengths) enter the detector at once. The detector registers an electric current having a height proportional to the photon energy, and these pulses are then separated electronically, using a pulse analyzer. In wavelength dispersive XRF spectrometers, the secondary x-rays are dispersed spatially by crystal diffraction on the basis of wavelength. The crystal and detector are made to synchronously rotate and the detector then receives only one wavelength at a time. The intensity of the x-rays emitted by each element is proportional to its concentration, after correcting for matrix effects. For nickel compounds and total chlorides, the XRF instrument response is expected to be linear to analyte concentration. Performance specifications and test procedures are provided to ensure reliable data.
3.0 Definitions.
3.1 Measurement System. The total equipment required for the determination of analyte concentration. The measurement system consists of the following major subsystems:
3.1.1 Sample Preparation. That portion of a system used for one or more of the following: sample acquisition, sample transport, sample conditioning, or sample preparation prior to introducing the sample into the analyzer.
3.1.2 Analyzer. That portion of the system that senses the analyte to be measured and generates an output proportional to its concentration.
3.1.3 Data Recorder. A digital recorder or personal computer used for recording measurement data from the analyzer output.
3.2 Span. The upper limit of the gas concentration measurement range displayed on the data recorder.
3.3 Calibration Standards. Prepared catalyst samples or other samples of known analyte concentrations used to calibrate the analyzer and to assess calibration drift.
3.4 Energy Calibration Standard. Calibration standard, generally provided by the XRF instrument manufacturer, used for assuring accuracy of the energy scale.
3.5 Accuracy Assessment Standard. Prepared catalyst sample or other sample of known analyte concentrations used to assess analyzer accuracy error.
3.6 Zero Drift. The difference in the measurement system output reading from the initial value for zero concentration level calibration standard after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
3.7 Calibration Drift. The difference in the measurement system output reading from the initial value for the mid-range calibration standard after a stated period of operation during which no unscheduled maintenance, repair, or adjustment took place.
3.8 Spectral Interferences. Analytical interferences and excessive biases caused by elemental peak overlap, escape peak, and sum peak interferences between elements in the samples.
3.9 Calibration Curve. A graph or other systematic method of establishing the relationship between the analyzer response and the actual analyte concentration introduced to the analyzer.
3.10 Analyzer Accuracy Error. The difference in the measurement system output reading and the ideal value for the accuracy assessment standard.
4.0 Interferences.
4.1 Spectral interferences with analyte line intensity determination are accounted for within the method program. No action is required by the XRF operator once these interferences have been addressed within the method.
4.2 The X-ray production efficiency is affected by particle size for the very lightest elements. However, particulate matter (PM) 2.5 particle size effects are substantially <1 percent for most elements. The calibration standards should be prepared with material of similar particle size or be processed (ground) to produce material of similar particle size as the catalyst samples to be analyzed. No additional correction for particle size is performed. Alternatively, the sample can be fused in order to eliminate any potential particle size effects.
5.0 Safety.
5.1 Disclaimer. This method may involve hazardous materials, operations, and equipment. This test method may not address all of the safety problems associated with its use. It is the responsibility of the user of this test method to establish appropriate safety and health practices and determine the applicability of regulatory limitations prior to performing this test method.
5.2 X-ray Exposure. The XRF uses X-rays; XRF operators should follow instrument manufacturer's guidelines to protect from accidental exposure to X-rays when the instrument is in operation.
5.3 Beryllium Window. In most XRF units, a beryllium (Be) window is present to separate the sample chamber from the X-ray tube and detector. The window is very fragile and brittle. Do not allow sample or debris to fall onto the window, and avoid using compressed air to clean the window because it will cause the window to rupture. If the window should rupture, note that Be metal is poisonous. Use extreme caution when collecting pieces of Be and consult the instrument manufacturer for advice on cleanup of the broken window and replacement.
6.0 Equipment and Supplies.
6.1 Measurement System. Use any measurement system that meets the specifications of this method listed in section 13. The typical components of the measurement system are described below.
6.1.1 Sample Mixer/Mill. Stainless steel, or equivalent to grind/mix catalyst and binders, if used, to produce uniform particle samples.
6.1.2 Sample Press/Fluxer. Stainless steel, or equivalent to produce pellets of sufficient size to fill analyzer sample window, or alternatively, a fusion device capable of preparing a fused disk of sufficient size to fill analyzer sample window.
6.1.3 Analytical Balance. ±0.0001 gram accuracy for weighing prepared samples (pellets).
6.1.4 Analyzer. An XRF spectrometer to determine the analyte concentration in the prepared sample. The analyzer must meet the applicable performance specifications in section 13.
6.1.5 Data Recorder. A digital recorder or personal computer for recording measurement data. The data recorder resolution (i.e., readability) must be 0.5 percent of span. Alternatively, a digital or analog meter having a resolution of 0.5 percent of span may be used to obtain the analyzer responses and the readings may be recorded manually.
7.0 Reagents and Standards.
7.1 Calibration Standards. The calibration standards for the analyzer must be prepared catalyst samples or other material of similar particle size and matrix as the catalyst samples to be tested that have known concentrations of the analytes of interest. Preparation (grinding/milling/fusion) of the calibration standards should follow the same processes used to prepare the catalyst samples to be tested. The calibration standards values must be established as the average of a minimum of three analyses using an approved EPA or ASTM method with instrument analyzer calibrations traceable to the U.S. National Institute of Standards and Technology (NIST), if available. The maximum percent deviation of the triplicate calibration standard analyses should agree within 10 percent of the average value for the triplicate analysis (see Figure 1). If the calibration analyses do not meet this criteria, the calibration standards must be re-analyzed. If unacceptable variability persists, new calibration standards must be prepared. Approved methods for the calibration standard analyses include, but are not limited to, EPA Methods 6010B, 6020, 7520, or 7521 of SW-846.1 Use a minimum of four calibration standards as specified below (see Figure 1):
7.1.1 High-Range Calibration Standard. Concentration equivalent to 80 to 100 percent of the span. The concentration of the high-range calibration standard should exceed the maximum concentration anticipated in the catalyst samples.
7.1.2 Mid-Range Calibration Standard. Concentration equivalent to 40 to 60 percent of the span.
7.1.3 Low-Range Calibration Standard. Concentration equivalent to 1 to 20 percent of the span. The concentration of the low-range calibration standard should be selected so that it is less than either one-fourth of the applicable concentration limit or of the lowest concentration anticipated in the catalyst samples.
7.1.4 Zero Calibration Standard. Concentration of less than 0.25 percent of the span.
7.2 Accuracy Assessment Standard. Prepare an accuracy assessment standard and determine the ideal value for the accuracy assessment standard following the same procedures used to prepare and analyze the calibration standards as described in section 7.1. The maximum percent deviation of the triplicate accuracy assessment standard analyses should agree within 10 percent of the average value for the triplicate analysis (see Figure 1). The concentration equivalent of the accuracy assessment standard must be between 20 and 80 percent of the span.
7.3 Energy Calibration Standard. Generally, the energy calibration standard will be provided by the XRF instrument manufacturer for energy dispersive spectrometers. Energy calibration is performed using the manufacturer's recommended calibration standard and involves measurement of a specific energy line (based on the metal in the energy calibration standard). This is generally an automated procedure used to assure the accuracy of the energy scale. This calibration standard may not be applicable to all models of XRF spectrometers (particularly wavelength dispersive XRF spectrometers).
8.0 Sample Collection, Preservation, Transport, and Storage. [Reserved]
9.0 Quality Control.
9.1 Energy Calibration. For energy dispersive spectrometers, conduct the energy calibration by analyzing the energy calibration standard provided by the manufacturer. The energy calibration involves measurement of a specific energy line (based on the metal in the energy calibration standard) and then determination of the difference between the measured peak energy value and the ideal value. This analysis, if applicable, should be performed daily prior to any sample analyses to check the instrument's energy scale. This is generally an automated procedure and assures the accuracy of the energy scale. If the energy scale calibration process is not automated, follow the manufacturer's procedures to manually adjust the instrument, as necessary.
9.2 Zero Drift Test. Conduct the zero drift test by analyzing the analyte concentration output by the measurement system with the initial calibration value for the zero calibration standard (see Figure 2). This analysis should be performed with each set of samples analyzed.
9.3 Calibration Drift Test. Conduct the calibration drift test by analyzing the analyte concentration output by the measurement system with the initial calibration value for the mid-range calibration standard (see Figure 2). This analysis should be performed with each set of samples analyzed.
9.4 Analyzer Accuracy Test. Conduct the analyzer accuracy test by analyzing the accuracy assessment standard and comparing the value output by the measurement system with the ideal value for the accuracy assessment standard (see Figure 2). This analysis should be performed with each set of samples analyzed.
10.0 Calibration and Standardization.
10.1 Perform the initial calibration and set-up following the instrument manufacturer's instructions. These procedures should include, at a minimum, the major steps listed in sections 10.2 and 10.3. Subsequent calibrations are to be performed when either a quality assurance/quality control (QA/QC) limit listed in section 13 is exceeded or when there is a change in the excitation conditions, such as a change in the tube, detector, X-ray filters, or signal processor. Calibrations are typically valid for 6 months to 1 year.
10.2 Instrument Calibration. Calibration is performed initially with calibration standards of similar matrix and binders, if used, as the samples to be analyzed (see Figure 1).
10.3 Reference Peak Spectra. Acquisition of reference spectra is required only during the initial calibration. As long as no processing methods have changed, these peak shape references remain valid. This procedure consists of placing the standards in the instrument and acquiring individual elemental spectra that are stored in the method file with each of the analytical conditions. These reference spectra are used in the standard deconvolution of the unknown spectra.
11.0 Analytical Procedure.
11.1 Sample Preparation. Prepare catalyst samples using the same procedure used to prepare the calibration standards. Measure and record the weight of sample used. Measure and record the amount of binder, if any, used. Pellets or films must be of sufficient size to cover the analyzer sample window.
11.2 Sample Analyses. Place the prepared catalyst samples into the analyzer. Follow the manufacturer's instructions for analyzing the samples.
11.3 Record and Store Data. Use a digital recorder or personal computer to record and store results for each sample. Record any mechanical or software problems encountered during the analysis.
12.0 Data Analysis and Calculations.
Carry out the following calculations, retaining at least one extra significant figure beyond that of the acquired data. Round off figures after final calculation.
12.1 Drift. Calculate the zero and calibration drift for the tests described in sections 9.2 and 9.3 (see also Figure 2) as follows:

Where:
CurrentAnalyzerCal.Response = Instrument response for current QC sample analyses;
InitialCal.Response = Initial instrument response for calibration standard;
QC Value = QC metric (zero drift or calibration drift), percent of span;
Span = Span of the monitoring system.
12.2 Analyzer Accuracy. Calculate the analyzer accuracy error for the tests described in section 9.4 (see also Figure 2) as follows:
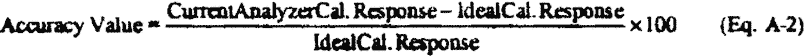
Where:
Accuracy Value = Percent difference of instrument response to the ideal response for the accuracy assessment standard;
CurrentAnalyzerCal.Response = Instrument response for current QC sample analyses;
IdealCal.Response = Ideal instrument response for the accuracy assessment standard.
13.0 Method Performance.
13.1 Analytical Range. The analytical range is determined by the instrument design. For this method, a portion of the analytical range is selected by choosing the span of the monitoring system. The span of the monitoring system must be selected such that it encompasses the range of concentrations anticipated to occur in the catalyst sample. If applicable, the span must be selected such that the analyte concentration equivalent to the emission standard is not less than 30 percent of the span. If the measured analyte concentration exceeds the concentration of the high-range calibration standard, the sample analysis is considered invalid. Additionally, if the measured analyte concentration is less than the concentration of the low-range calibration standard but above the detectable limit, the sample analysis results must be flagged with a footnote stating, in effect, that the analyte was detected but that the reported concentration is below the lower quantitation limit.
13.2 Minimum Detectable Limit. The minimum detectable limit depends on the signal-to-noise ratio of the measurement system. For a well-designed system, the minimum detectable limit should be less than 2 percent of the span.
13.3 Zero Drift. Less than ±2 percent of the span.
13.4 Calibration Drift. Less than ±5 percent of the span.
13.5 Analyzer Accuracy Error. Less than ±10 percent.
14.0 Pollution Prevention. [Reserved]
15.0 Waste Management. [Reserved]
16.0 Alternative Procedures. [Reserved]
17.0 References.
1. U.S. Environmental Protection Agency. 1998. Test Methods for Evaluating Solid Waste, Physical/Chemical Methods. EPA Publication No. SW-846, Revision 5 (April 1998). Office of Solid Waste, Washington, DC.
18.0 Tables, Diagrams, Flowcharts, and Validation Data.
Date: Analytic Method Used: Zeroa Low-Rangeb Mid-Rangec High-Ranged Accuracy Stde Sample Run: 1 2 3 Average Maximum Percent Deviation Figure 1. Data Recording Sheet for Analysis of Calibration Samples.
Source Identification:
Run Number:
Test Personnel:
Span:
Date:
Initial calibration response Current analyzer calibration response Drift (percent of span) Zero Standard Mid-range Standard Ideal calibration response Current analyzer calibration response Accuracy error (percent of ideal) Accuracy Standard Figure 2. Data Recording Sheet for System Calibration Drift Data.
[70 FR 6970, Feb. 9, 2005, as amended at 80 FR 75325, Dec. 1, 2015]