-
Start Preamble
Start Printed Page 6204
AGENCY:
Food and Drug Administration, HHS.
ACTION:
Proposed rule.
SUMMARY:
The Food and Drug Administration (FDA or Agency) is issuing this proposed rule to put into effect a final monograph for nonprescription, over-the-counter (OTC) sunscreen drug products. This proposed rule describes the conditions under which FDA proposes that OTC sunscreen monograph products are generally recognized as safe and effective (GRASE) and not misbranded. It is being published as part of the ongoing review of OTC drug products conducted by FDA. It is also being published to comply with the Federal Food, Drug, and Cosmetic Act (FD&C Act), as amended by the Sunscreen Innovation Act (SIA).
DATES:
Submit either electronic or written comments. on the proposed rule by May 28, 2019. Electronic comments must be submitted on or before May 28, 2019. The https://www.regulations.gov electronic filing system will accept comments until 11:59 p.m. Eastern Time at the end of May 28, 2019. See section XII for proposed effective and compliance dates of a final rule based on this document.
ADDRESSES:
You may submit comments as follows. Please note that late, untimely filed comments will not be considered. Comments received by mail/hand delivery/courier (for written/paper submissions) will be considered timely if they are postmarked or the delivery service acceptance receipt is on or before the closing date.
Please be advised that safety and effectiveness data that are not available to the public cannot be relied on to establish conditions under which the OTC drugs described in this document of proposed rulemaking are generally recognized as safe and effective. Accordingly, you should not submit, and FDA generally does not intend to rely on, any evidence of safety and effectiveness that bears a confidential mark unless you include a statement that the information may be released to the public. Similarly, if your submission includes safety and effectiveness data or information marked as confidential by a third party (such as a contract research organization or consultant), you should either include a statement that you are authorized to make the information publicly available or include an authorization from the third party permitting the information to be publicly disclosed. If you submit data without confidential markings in response to this document and such data includes studies or other information that were previously submitted confidentially (e.g., as part of a new drug application), FDA intends to presume that you intend to make such data publicly available.
Electronic Submissions
Submit electronic comments in the following way:
- Federal eRulemaking Portal: https://www.regulations.gov. Follow the instructions for submitting comments. Comments submitted electronically, including attachments, to https://www.regulations.gov will be posted to the docket unchanged. Because your comment will be made public, you are solely responsible for ensuring that your comment does not include any confidential information that you or a third party may not wish to be posted, such as medical information, your or anyone else's Social Security number, or confidential business information, such as a manufacturing process. Please note that if you include your name, contact information, or other information that identifies you in the body of your comments, that information will be posted on https://www.regulations.gov.
- If you want to submit a comment with confidential information that you do not wish to be made available to the public, submit the comment as a written/paper submission and in the manner detailed (see “Written/Paper Submissions” and “Instructions”).
Written/Paper Submissions
Submit written/paper submissions as follows:
- Mail/Hand Delivery/Courier (for written/paper submissions): Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
- For written/paper comments submitted to the Dockets Management Staff, FDA will post your comment, as well as any attachments, except for information submitted, marked and identified, as confidential, if submitted as detailed in “Instructions.”
Instructions: All submissions received must include the Docket No. FDA-1978-N-0018 (formerly Docket No. FDA-1978-N-0038) for “Sunscreen Drug Products for Over-the-Counter Human Use.” Received comments, those filed in a timely manner (see ADDRESSES), will be placed in the docket and, except for those submitted as “Confidential Submissions,” publicly viewable at https://www.regulations.gov or at the Dockets Management Staff between 9 a.m. and 4 p.m., Monday through Friday.
- Confidential Submissions—To submit a comment with confidential information that you do not wish to be made publicly available, submit your comments only as a written/paper submission. You should submit two copies total. One copy will include the information you claim to be confidential with a heading or cover note that states “THIS DOCUMENT CONTAINS CONFIDENTIAL INFORMATION.” The Agency will review this copy, including the claimed confidential information, in its consideration of comments. The second copy, which will have the claimed confidential information redacted/blacked out, will be available for public viewing and posted on https://www.regulations.gov. Submit both copies to the Dockets Management Staff. If you do not wish your name and contact information to be made publicly available, you can provide this information on the cover sheet and not in the body of your comments and you must identify this information as “confidential.” Any information marked as “confidential” will not be disclosed except in accordance with 21 CFR 10.20 and other applicable disclosure law. For more information about FDA's posting of comments to public dockets, see 80 FR 56469, September 18, 2015, or access the information at: https://www.gpo.gov/fdsys/pkg/FR-2015-09-18/pdf/2015-23389.pdf.
Docket: For access to the docket to read background documents or the electronic and written/paper comments received, go to https://www.regulations.gov and insert the docket number, found in brackets in the heading of this document, into the “Search” box and follow the prompts and/or go to the Dockets Management Staff, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
Submit comments on information collection issues under the Paperwork Reduction Act of 1995 to the Office of Management and Budget (OMB) in the following ways:
- Fax to the Office of Information and Regulatory Affairs, OMB, Attn: FDA Desk Officer, Fax: 202-395-7285, or email to oira_submission@omb.eop.gov. All comments should be identified with Start Printed Page 6205the title, “Sunscreen Drug Products for Over-the-Counter Human Use.”
The Agency encourages commenters also to submit their comments on these paperwork requirements to the rulemaking docket (Docket No. FDA-1978-N-0018), along with their comments on other parts of the proposed rule.
Start Further InfoFOR FURTHER INFORMATION CONTACT:
Kristen Hardin, Center for Drug Evaluation and Research, Food and Drug Administration, 10903 New Hampshire Ave., Bldg. 22, Rm. 5443, Silver Spring, MD 20993, 240-402-4246.
End Further Info End Preamble Start Supplemental InformationSUPPLEMENTARY INFORMATION:
Table of Contents
I. Executive Summary
A. Purpose and Coverage of the Proposed Rule
B. Summary of the Major Provisions of the Proposed Rule
C. Legal Authority
D. Costs and Benefits
II. Table of Abbreviations/Commonly Used Acronyms in This Document
III. Background
A. FDA's Current Regulatory Framework
B. History of This Rulemaking
IV. Scope of This Rulemaking
V. Legal Authority
VI. Need for Additional Safety Information
A. Increased Consumer Exposure to Sunscreen Active Ingredients
B. Emerging Safety Concerns
VII. Framework for Evaluation of Safety Data
A. General
B. Clinical Safety Testing
C. Nonclinical Safety Testing
D. Postmarketing Safety Data
E. Sunscreens Containing Nanomaterials
VIII. Existing Safety Data for Sunscreen Active Ingredients
A. Ingredients Proposed as Category I
B. Ingredients Proposed as Category II
C. Ingredients Proposed as Category III
D. Anticipated Final Formulation In Vitro Permeation Testing
IX. Additional Proposed Conditions of Use
A. Proposed Requirements Related to Dosage Form
B. Proposed Maximum SPF and Broad Spectrum Requirements
C. Proposed PDP Labeling Requirements
D. Proposed Requirements Related to Final Formulation Testing and Recordkeeping
E. Proposed Status of Sunscreen-Insect Repellent Combination Products
X. Proposed Actions To Effectuate Lifting of Stay and Harmonize Impacted Regulations
XI. Comment Period
XII. Proposed Effective/Compliance Dates
XIII. Preliminary Economic Analysis of Impacts
A. Introduction
B. Summary of Costs and Benefits
XIV. Analysis of Environmental Impact
XV. Paperwork Reduction Act of 1995
A. Labeling for Sunscreen Products and Associated Clinical Testing
B. Regulatory Status of Testing Entities
C. Generating and Maintaining Records of SPF and Broad Spectrum Testing
XVI. Federalism
XVII. Consultation and Coordination With Indian Tribal Governments
XVIII. References
I. Executive Summary
A. Purpose and Coverage of the Proposed Rule
The Food and Drug Administration (FDA or Agency) is publishing this proposed rule as part of the regulatory proceeding to put into effect a final monograph [1] for nonprescription, OTC sunscreen drug products under the OTC Drug Review. In 2011, FDA announced that “we are considering certain active ingredient safety issues further . . . In a forthcoming rulemaking, we intend to request additional data regarding the safety of the individual sunscreen active ingredients.” (“Revised Effectiveness Determination; Sunscreen Drug Products for Over-the-Counter Human Use” (Max SPF PR), 76 FR 35672 at 35673, June 17, 2011). As described in further detail below, changed conditions in the nearly 20 years since publication of the final rule “Sunscreen Drug Products for Over the Counter Human Use” (64 FR 27666, May 21, 1999) (now stayed) (Stayed 1999 Final Monograph) have meant that additional safety data are now needed to establish that certain of the active ingredients listed in the Stayed 1999 Final Monograph are GRASE for use in sunscreen products.[2]
As detailed below, we emphasize that this proposed rule does not represent a conclusion by FDA that the sunscreen active ingredients included in the Stayed 1999 Final Monograph but proposed here as Category III are unsafe for use in sunscreens. Rather, we are requesting additional information on these ingredients so that we can evaluate their GRASE status in light of changed conditions, including substantially increased sunscreen usage and exposure and evolving information about the potential risks associated with these products since they were originally evaluated. While these additional data are being developed and reviewed, FDA generally intends to follow the enforcement approach discussed in section III.B with regard to sunscreen products that contain those sunscreen active ingredients included in the Stayed 1999 Final Monograph.
This proposed rule is also being published to comply with section 586E of the FD&C Act (21 U.S.C. 360fff-5), as amended by the SIA (21 U.S.C. ch. 9, sub. 5, part I, enacted November 26, 2014). The SIA calls for FDA to issue a final OTC sunscreen monograph to be effective within 5 years of enactment of the SIA, or by November 26, 2019 (section 586E(a) of the FD&C Act). If the final OTC sunscreen monograph does not include provisions related to the effectiveness of various sun protection factor (SPF) levels and address all dosage forms known to FDA to be used in sunscreens marketed in the United States without approved new drug applications (NDAs), the SIA requires FDA, among other things, to submit a report to Congress explaining these omissions (section 586E(b) of the FD&C Act). As explained in section I.B, in this proposed rule, FDA is addressing multiple conditions of use applicable to sunscreen monograph products, including both the effectiveness of various SPF values and all marketed sunscreen dosage forms (and intends to do so in the final rule as well).
This proposed rule does not address the sunscreen active ingredients that were originally submitted under the procedures established in FDA's time and extent application (TEA) regulation (§ 330.14 (21 CFR 330.14)) (67 FR 3074, January 23, 2002), and are now being addressed through a process set forth in the SIA.
B. Summary of the Major Provisions of the Proposed Rule
1. Proposed GRASE Status of Active Ingredients Listed in the Stayed 1999 Final Monograph
a. Framework for evaluation of safety data. As previously noted, changed conditions in the time since issuance of the Stayed 1999 Final Monograph have meant that additional safety data are now needed to establish that certain of the active ingredients listed in the Stayed 1999 Final Monograph are GRASE for use in sunscreen products in accordance with the standards established in § 330.10(a)(4) (21 CFR Start Printed Page 6206330.10(a)(4)). FDA's approach to the clinical safety evaluation of OTC sunscreen active ingredients is based on our current scientific understanding regarding the safety evaluation of topical drug products for chronic use, and is therefore generally consistent with the safety data needed to meet the requirements for approval of an NDA for a chronic-use topical drug product (e.g., topical safety studies (irritation, sensitization, and photosafety); bioavailability (absorption); and evaluation of adverse events observed in clinical studies). Postmarketing safety information is also relevant to our safety evaluation.
Our current approach to the nonclinical safety evaluation of these active ingredients takes into account their lengthy marketing history in the United States. Unlike the nonclinical data required to meet the standard for approval of chronic-use topical NDA products (which include comprehensive nonclinical pharmacology and toxicology safety testing), the approach to nonclinical safety testing reflected in this proposed rule is largely focused on potential long-term adverse effects or effects not otherwise readily detected from human use (i.e., carcinogenicity and reproductive toxicity).
b. Existing safety data for ingredients listed in Stayed 1999 Final Monograph. In section VIII, we discuss our review of the scientific literature, submissions to the sunscreen monograph docket, and adverse event reports submitted to FDA's Adverse Event Reporting System (FAERS) for the ingredients listed in the Stayed 1999 Final Monograph and identify any existing gaps. Because our review of this evidence has produced sufficient safety data on both zinc oxide and titanium dioxide to support a proposal that sunscreen products containing these ingredients (at concentrations of up to 25 percent) would be GRASE, we are proposing that these ingredients are Category I. Our evaluation of the available safety data for aminobenzoic acid (PABA) and trolamine salicylate, however, has caused us to conclude that the risks associated with use of these active ingredients in sunscreen products outweigh their benefits. In the case of trolamine salicylate, these risks include the potential for serious detrimental health effects (including bleeding) caused by the anti-coagulation effects of salicylic acid and increased risk of salicylate toxicity when this ingredient is used in sunscreens. For PABA, the risks include significant rates of allergic and photoallergic skin reactions, as well as cross-sensitization with structurally similar compounds. Accordingly, we are proposing that these two ingredients are Category II.
Because the public record does not currently contain sufficient data to support positive GRASE determinations for cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, sulisobenzone, oxybenzone, or avobenzone, we are proposing that these ingredients are Category III. For example, the available literature includes studies indicating that oxybenzone is absorbed through the skin to a greater extent than previously understood and can lead to significant systemic exposure, as well as data showing the presence of oxybenzone in human breast milk, amniotic fluid, urine, and blood plasma. The significant systemic availability of oxybenzone, coupled with a lack of data evaluating the full extent of its absorption potential, is a concern, among other reasons, because of questions raised in the published literature regarding the potential for endocrine activity in connection with systemic oxybenzone exposure. Nearly all of these sunscreen active ingredients also have limited or no data characterizing their absorption.
2. Proposed Requirements Related to Dosage Forms
In 2011, FDA published an Advance Notice of Proposed Rulemaking (ANPR) that identified sunscreen dosage forms considered either eligible or ineligible for inclusion in the sunscreen monograph, and specifically requested comments on the safety and efficacy of spray sunscreens. After considering comments received in response (and other available data), we are proposing the following dosage forms as Category I: Oils, lotions, creams, gels, butters, pastes, ointments, and sticks. We are also proposing Category I status for spray sunscreens, subject to testing necessary to minimize potential risks from unintended inhalation (particle size restrictions) and flammability (flammability and drying time testing), together with related labeling requirements. We are proposing to add sunscreen powders to the list of those eligible for inclusion in the monograph and proposing that this dosage form is Category III; we expect that powders would also be subject to particle size restrictions if found to be GRASE in the final monograph. Finally, we are proposing that sunscreens in all other dosage forms—including wipes, towelettes, body washes, and shampoos—are new drugs because we did not receive data showing that they were marketed prior to 1972, as required for inclusion in the monograph.
3. Proposed Maximum Sun Protection Factor and Broad Spectrum Requirements
In the Stayed 1999 Final Monograph, FDA established SPF 30+ as the maximum labeled SPF value for sunscreen monograph products, and subsequently proposed (in 2011) to raise this value to SPF 50+. Because of evidence showing additional meaningful clinical benefit associated with broad spectrum sunscreen products with an SPF of 60, we are now proposing to raise the maximum labeled SPF value to SPF 60+. Given the lack of data showing that sunscreens with SPF values above 60 provide additional meaningful clinical benefit, we are proposing not to allow labeled SPF values higher than 60+.
While our proposed cap for SPF labeling is SPF 60+, we are proposing to permit the marketing of sunscreen products formulated with SPF values up to 80. This formulation margin is intended to provide manufacturers with formulation flexibility that we hope will: (1) Help facilitate the development of products with greater Ultraviolet A (UVA) protection and (2) more fully account for the range of variability in SPF test results (discussed further in sections IX.B.4.b-c) for sunscreen products labeled SPF 60+. We are proposing not to allow the marketing (without an approved NDA) of sunscreen products with SPF values above SPF 80.
In addition, since publication of the 2011 “Labeling and Effectiveness Testing; Sunscreen Drug Products for Over-the-Counter Human Use” (L&E Final Rule) (76 FR 35620, June 17, 2011) and Max SPF PR, the body of scientific evidence linking UVA exposure to skin cancers and other harms has grown significantly. This evidence raises concerns about the potential for inadequate UVA protection in marketed sunscreen products—particularly in high SPF sunscreen products that either do not pass the current broad spectrum test or (though they pass our current broad spectrum test) have inadequate uniformity in their UVA protection. Consumers using these products may, while successfully preventing sunburn, accumulate excessively large doses of UVA radiation—thereby exposing themselves to additional risks related to skin cancer and early skin aging.
To address these concerns, we are making a number of proposals designed to couple a greater magnitude of UVA protection to increases in SPF values. We are proposing to require that all sunscreen products with SPF values of 15 and above satisfy broad spectrum Start Printed Page 6207requirements. Among other things, this proposal eliminates the potential confusion permitted by the current labeling regime, in which a higher numbered product (for example, one labeled SPF 30) may provide inferior protection against UVA radiation than a lower numbered product (for example, one labeled broad spectrum SPF 15). We are also proposing to add to the current broad spectrum test a requirement that broad spectrum products meet a UVA I/UV ratio of 0.7 or higher. Given how much of the UVA portion of the ultraviolet (UV) spectrum is composed of UVA I radiation, and given what we now know about the skin cancer risks associated with UVA exposure, ensuring that sunscreen products provide adequate protection in the UVA I portion of the spectrum is critical.[3] Because sunscreens with SPF 2 to 14 have not been demonstrated to help reduce the risk of skin cancer and early skin aging caused by the sun, whether or not they provide protection against UVA radiation as well as ultraviolet B (UVB) radiation, we are not proposing to require that they pass the revised broad spectrum test. However, we seek comment on whether these low SPF products should remain in the market.
Finally, we are proposing to require that sunscreen products with SPF values of 15 or above be labeled with an SPF number corresponding to the lowest number in a range of tested SPF results. For example, sunscreens testing at SPF 15-19 would be labeled “SPF 15”; those testing at 40-49 would be labeled “SPF 40.” We are making this proposal because new evidence has caused us to reexamine the variability inherent in the SPF test (which relies on visual assessments of erythema in human subjects). The data we reviewed suggests that the clinical evaluation undertaken during SPF testing creates variability that justifies the use of SPF ranges. As explained further in sections IX.B.4.b-c, because this variability is exacerbated at high SPFs, we are proposing that sunscreens testing at SPF 30 or more be labeled in increments of 10 (i.e., SPF 30, SPF 40, SPF 50, with a proposed maximum of SPF 60+), that sunscreens testing at SPF 15 to 29 be labeled in increments of 5 (i.e., SPF 15, SPF 20, SPF 25), and that the requirement that labeled SPF values correspond to ranges (rather than precise numerical values) is not necessary below SPF 15.
4. Proposed PDP Labeling Requirements
We are also proposing to partially revise the current requirements for information that must appear on the principal display panel (PDP) of sunscreen products. The PDP is the part of a product label that is most likely to be viewed or examined when the product is displayed for retail sale. A major feature of the PDP is the statement of identity (SOI). We are proposing that the SOI consist of an alphabetical listing of the sunscreen active ingredients in the product, followed by “Sunscreen” and the product's dosage form (such as lotion or spray). This information would supplement other important elements of the PDP (e.g., SPF, broad spectrum, and water resistance information) to provide a succinct summary of the product's key characteristics on the front of the package or container, permitting consumers to more readily compare products and either select or avoid a given product accordingly. For sunscreen products that have not been shown to help prevent skin cancer or early skin aging caused by the sun, the SPF statement would be followed by an asterisk (*) directing consumers to see the “Skin Cancer/Skin Aging alert” elsewhere on the label. Finally, to prevent required information from being obscured or overwhelmed by other labeling features, we are revising the format requirements for the SPF, broad spectrum, and water resistance statements on the PDP.
5. Proposed Requirements Related to Final Formulation Testing Processes and Recordkeeping
To ensure that FDA can assess compliance with our regulations, we are proposing to require records of required final formulation testing of sunscreen products to be maintained for 1 year after the product expiration date, or, if the product is exempt from expiration dating (as most sunscreens are), for 3 years after distribution of the last lot labeled in reliance on that testing. In addition, we are proposing to require responsible persons (defined in section IX.D.2.b) to keep records of sunscreen formulation testing, and clarifying that required records would be subject to FDA inspection. We are also proposing a number of revisions to our labeling and testing regulations designed to clarify FDA expectations about clinical final formulation testing processes and to ensure that the testing of marketed sunscreen products is conducted in a manner that both protects human subjects and produces reliable results.
6. Proposed Status of Sunscreen-Insect Repellent Combination Products
The proposed rule also addresses sunscreen-insect repellent products, which are jointly regulated by FDA as sunscreen drugs and by the Environmental Protection Agency (EPA) as pesticides under the Federal Insecticide, Fungicide and Rodenticide Act (FIFRA). In 2007, FDA and EPA both issued ANPRs requesting comment on the appropriate regulatory status of these products. We are proposing to classify these products as Category II because incompatibilities between FDA and EPA labeling requirements prevent these products from being labeled in a manner that sufficiently ensures safe and effective use of the sunscreen component and provides adequate directions for use. In addition, there are data suggesting that combining some sunscreen active ingredients with the insecticide DEET may increase absorption of either or both components.
7. Proposed Actions To Effectuate Lifting of Stay and Harmonize Impacted Regulations
Finally, we are proposing to lift the stay on the 1999 Final Monograph (subject to the revisions to parts 201, 310, 347, and 352 (21 CFR parts 201,[4] 310, 347, and 352) described in this document), and have proposed revisions to these regulations necessary to effectuate the lifting of the stay and to harmonize any impacted regulations.
C. Legal Authority
We are issuing this proposed rule under sections 201, 301, 501, 502, 503, 505, 510, 586E, 701, 702, 703, 704, and 721 of the FD&C Act (21 U.S.C. 321, 331, 351, 352, 353, 355, 360, 360fff-5, 371, 372, 373, 374, and 379e) and under section 351 of the Public Health Service Act (42 U.S.C. 262).
D. Costs and Benefits
If finalized, the proposed rule would update and make effective regulations to ensure the safety and effectiveness of sunscreen products marketed under the OTC drug monograph. The rule would update sunscreen product labeling standards, address the safety of sunscreen active ingredients, revise and Start Printed Page 6208clarify our expectations for testing and recordkeeping by entities that conduct sunscreen testing, and address other sunscreen safety or efficacy concerns, like combination sunscreen-insect repellents and alternative dosage forms.
Consumers would benefit from less exposure to sunscreen products containing active ingredients about which safety questions remain, less exposure to sunscreen products labeled with potentially misleading sun protection information, increased consumption of products with better UVA protection, less exposure to flammable spray sunscreens, and less exposure to spray and powder sunscreen products posing inhalation risks. Consumers would also experience transaction cost savings. The costs of the rule to sunscreen manufacturers include administrative costs, costs to fill data gaps for active ingredients and powder dosage forms, product formulation testing costs, and costs to reformulate and relabel sunscreen products. Finally, testing entities would incur recordkeeping costs if they do not already maintain adequate records of testing equipment, methods, and observations in final formulation testing.
II. Table of Abbreviations/Commonly Used Acronyms in This Document
Abbreviation/ acronym What it means ANDA Abbreviated new drug application. ANPR Advance notice of proposed rulemaking. CFR Code of Federal Regulations. DART Developmental and reproductive toxicity. DEET N,N-Diethyl-meta-toluamide. EPA Environmental Protection Agency. FAERS FDA's Adverse Event Reporting System. FDA or Agency Food and Drug Administration. FD&C Act Federal Food, Drug, and Cosmetic Act. FIFRA Federal Insecticide, Fungicide, and Rodenticide Act. FR Federal Register. GRASE Generally recognized as safe and effective (or general recognition of safety and effectiveness). ICH International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. IND Investigational new drug application. IRB Institutional Review Board. mL Milliliter. MUsT Maximal usage trial. NDA New drug application. NDAC Nonprescription Drugs Advisory Committee. Ng Nanogram. Nm Nanometer. NOAEL No observed adverse effect level. NPIC National Pesticide Information Center. NTP National Toxicology Program of the National Institutes of Health. OMB Office of Management and Budget. OTC Over-the-counter. PABA Aminobenzoic acid. ROS Reactive oxygen species. SIA Sunscreen Innovation Act. SPF Sun protection factor. TEA Time and extent application. TFM Tentative final monograph. U.S.C. United States Code. USP United States Pharmacopeia. UVA Ultraviolet A. UVB Ultraviolet B. III. Background
A. FDA's Current Regulatory Framework
In the following sections, we provide a brief description of terminology used in the OTC Drug Review regulations as well as an overview of OTC sunscreen products, their intended uses, and FDA's regulation of them.
1. Terminology
a. OTC drug review. The OTC Drug Review is the process established by FDA to evaluate the safety and effectiveness of OTC drug products marketed in the United States before May 11, 1972, and to establish the conditions under which they are considered to be GRASE and not misbranded. As described further below, the OTC Drug Review is generally conducted via a multiphase public rulemaking process (each phase requiring a Federal Register publication), resulting in the establishment of a monograph for an OTC therapeutic drug category.
b. Generally recognized as safe and effective (GRASE). An OTC drug is “generally recognized as safe and effective” if it meets each of the conditions contained in an applicable OTC final monograph, the conditions contained in part 330 (21 CFR part 330), and any other applicable regulatory and statutory requirements for OTC drugs, including the labeling requirements in part 201.
c. Proposed, tentative final, and final monographs. The proposed monograph, which is typically published in the form of an ANPR, is the end product of the first phase of the rulemaking process described above. After reviewing the report and recommendations of an expert advisory review panel responsible for initially reviewing the safety, effectiveness, and labeling of products in a given therapeutic category, FDA publishes a proposed monograph (together with the report and recommendations of the expert review panel) (see § 330.10(a)(6)). After a period of public comment, FDA publishes a tentative final monograph (TFM) (in the form of a proposed rule, proposing conditions under which OTC drugs in the therapeutic class being considered are GRASE and not misbranded (see § 330.10(a)(7)). Following public comment on the TFM, FDA publishes a final monograph in FDA's regulations (see 21 CFR chapter I, subchapter D) codifying the conditions under which products in the OTC therapeutic drug category are GRASE and not misbranded (see § 330.10(a)(9)). An OTC drug may be legally marketed without an approved NDA or abbreviated new drug application (ANDA) if it meets each of the conditions contained in an applicable final monograph, the conditions contained in part 330, and any other applicable regulatory and statutory requirements for OTC drugs, including the labeling requirements in part 201.
d. Category I, II, and III classifications. In the course of establishing an OTC monograph, active ingredients and other OTC drug conditions are classified in one of three categories: Category I (conditions under which a nonprescription drug in the therapeutic category would be GRASE and not misbranded), Category II (conditions that would result in the drug being classified as not GRASE and/or misbranded) and Category III (conditions proposed to be excluded from the final monograph because available data are insufficient to classify them as either Category I or Category II) (see § 330.10(a)(6)).
2. OTC Sunscreen Products Regulated Under the OTC Drug Review and Their Intended Uses
OTC sunscreen drugs regulated under the OTC Drug Review are topically applied products indicated to help prevent sunburn; some are also indicated to decrease the risk of skin cancer and early skin aging caused by exposure to the sun's UV radiation (when used as directed with other sun protection measures) (see § 201.327(c)). The active ingredients in sunscreen products achieve these protective effects by absorbing, reflecting, and/or scattering radiation in the UV range (from 290 to 400 nanometers (nm)) (see section 586(10) of the FD&C Act (21 U.S.C. 360fff(10)); see also § 352.3(c) (21 CFR 352.3(c)), stayed).
Sunscreen products must be labeled with an SPF value calculated using a standardized SPF testing procedure set forth in FDA regulations (in § 201.327(i)). As discussed in further detail in section IX.B.1, the SPF test Start Printed Page 6209measures the amount of UV radiation exposure it takes to cause sunburn when a person is using a sunscreen when compared with how much UV exposure it takes to cause sunburn when the person is not using a sunscreen. Because SPF values represent a sunscreen's level of sunburn protection, they are primarily (though not exclusively) an indicator of expected protection from UVB radiation (see section IX.B.1 for a discussion of both UVB and UVA radiation).
To pass FDA's current test for the inclusion of the term “broad spectrum” in labeling (which was established in the 2011 L&E Final Rule), sunscreen products must demonstrate that, in addition to UVB protection, they also provide UVA protection. Further, only products that have been demonstrated both to provide broad spectrum protection and to have a minimum SPF value of 15 have been shown to reduce the risk of skin cancer and early skin aging caused by the sun (when used as directed with other sun protection measures). By contrast, sunscreens that have not been demonstrated to provide both broad spectrum protection and an SPF value of at least 15 have only been demonstrated to help prevent sunburn.[5] Thus, under the 2011 L&E Final Rule, passing the broad spectrum test in § 201.327(j) (21 CFR 201.327(j)) is necessary, but not itself sufficient, to support inclusion of a skin cancer indication in labeling, although any product that passes the broad spectrum test may be labeled with the term “Broad Spectrum” in conjunction with its SPF value.
B. History of This Rulemaking
1. The OTC Sunscreen Drug Review and FDA's Regulation of OTC Sunscreen Drug Products
Our initial call for safety and efficacy data for sunscreen products was issued in 1972 (37 FR 26456, December 12, 1972). The resulting data submissions were reviewed by the Advisory Review Panel on OTC Topical Analgesic, Antirheumatic, Otic, Burn, and Sunburn Prevention and Treatment Products, whose panel report and recommended monograph were published as an ANPR in 1978 (43 FR 38206, August 25, 1978). The ANPR contained a list of the 21 sunscreen active ingredients [6] that the panel recommended for classification as GRASE when used under the conditions described in the panel's report (43 FR 38206 at 38219). In 1993, having reviewed the panel's report and related public comments, FDA published a TFM (58 FR 28194, May 12, 1993) which (with one exception—padimate A) proposed as GRASE all of the active ingredients that had been included in the ANPR. The TFM also included specified maximum concentrations at which the proposed ingredients would be considered GRASE for use in sunscreens.
In the years following the publication of the 1993 TFM, FDA removed several additional ingredients from the TFM (see 59 FR 29706, June 8, 1994), as described at 64 FR 27666 at 27681, and proposed the inclusion of two more.[7] In 1999, FDA published a final sunscreen monograph, which included the following 16 sunscreen active ingredients along with the conditions (including maximum concentrations) under which these ingredients would be considered GRASE for use in sunscreens: [8]
Table 1—Sunscreen Active Ingredients Included in the Stayed 1999 Final Monograph
Active ingredient Maximum concentration (%) Aminobenzoic acid (PABA) 15 Avobenzone 3 Cinoxate 3 Dioxybenzone 3 Ensulizole 4 Homosalate 15 Meradimate 5 Octinoxate 7.5 Octisalate 5 Octocrylene 10 Oxybenzone 6 Padimate O 8 Sulisobenzone 10 Titanium dioxide 25 Trolamine salicylate 12 Zinc oxide 25 Among other things, the Stayed 1999 Final Monograph established a minimum SPF value of 2, and an SPF of 30+ as the maximum labeled SPF value (64 FR 27666). FDA concluded that the above-listed ingredients (at the listed concentrations) could also be used in combination, with limited exceptions, provided that each active ingredient contributed a minimum SPF of 2 to the finished product (64 FR 27666).[9]
The effective date for complying with the Stayed 1999 Final Monograph was May 21, 2001. This deadline was extended (65 FR 36319, June 8, 2000) and then stayed until further notice (66 FR 67485, December 31, 2001) to provide additional time to resolve various outstanding issues, such as the labeling and testing of finished OTC sunscreen products. As a result, the Start Printed Page 6210Stayed 1999 Final Monograph has never been in effect.
In 2011, FDA published a draft guidance for industry, “Enforcement Policy—OTC Sunscreen Drug Products Marketed Without an Approved Application,” addressing the circumstances under which FDA intended to exercise its enforcement discretion with respect to certain marketed OTC sunscreen products in the period until a final OTC sunscreen monograph becomes effective. This guidance was finalized in May 2018 (2018 Final Guidance) (Ref. 1). Unless the failure to pursue regulatory action poses a potential health hazard to the consumer, FDA generally does not intend to object to the marketing of OTC sunscreen products that do not have an approved NDA or ANDA provided that they: (1) Contain as sunscreen active ingredients only the active ingredients or combinations of active ingredients listed in 21 CFR 352.10 and 352.20 (both currently stayed); (2) do not make claims addressed in §§ 201.327(c)(3) and (g) and 310.545(a)(29)(ii); (3) comply with the requirements for OTC drugs set forth in part 201 and § 330.1 (21 CFR 330.1), the requirements for adverse event reporting for OTC drugs set forth in the FD&C Act (see section 760 (21 U.S.C. 379aa)), and the provisions of the FD&C Act addressing adulteration; and (4) follow applicable labeling and testing requirements for OTC sunscreens set forth in § 201.327.
2. Recent Significant Rulemakings Relevant to This Proposed Rule
Since publishing the Stayed 1999 Final Monograph, FDA has issued a number of Federal Register notices relating to OTC sunscreens. Major notices pertinent to today's proposed rule are summarized briefly in table 2 below:
Table 2—Recent Significant Federal Register Notices Pertinent to This Rule
Federal Register notice Information in notice Insect Repellent-Sunscreen Drug Products for Over-the-Counter Human Use: Request for Information and Comments; 72 FR 7941, February 22, 2007 We issued a notice stating that we were considering amending the Stayed 1999 Final Monograph to include conditions for marketing insect repellent-sunscreen drug products and requested information to form a regulatory position on these products. The Environmental Protection Agency, which regulates the insect repellent component of insect repellent-sunscreen combinations, published a similar notice concurrently with ours, also seeking information and comment on these products. Sunscreen Drug Products for Over-the-Counter Human Use: Proposed Amendment of Final Monograph; 72 FR 49070, August 27, 2007 We proposed to amend the Stayed 1999 Final Monograph to address, among other things, formulation, labeling, and testing requirements for both UVA and UVB radiation protection. Labeling and Effectiveness Testing: Sunscreen Drug Products for Over-the-Counter Human Use (L&E Final Rule); 76 FR 35620, June 17, 2011 We issued a final rule establishing labeling and testing requirements for sunscreen products. Among other things, the L&E Final Rule established optional broad spectrum labeling, created an optional indication relating to decreasing the risk of skin cancer and early skin aging for broad spectrum products with an SPF of 15 or higher, and required a labeling warning for sunscreens that did not both satisfy the broad spectrum test and provide an SPF of at least 15. 2011 Proposed Rule: Revised Effectiveness Determination (Max SPF PR); 76 FR 35672, June 17, 2011 We proposed to raise the limit on the maximum permissible labeled SPF value for sunscreen products to “50+.” Among other things, we sought comment on the appropriateness of a formulation cap for sunscreen products. 2011 ANPR and Request for Data and Information on Certain Dosage Forms; 76 FR 35669, June 17, 2011 We issued an ANPR describing the sunscreen dosage forms that we considered to be part of the OTC Drug Review and thus eligible for potential inclusion in a sunscreen monograph, as well as those dosage forms that we did not consider eligible. We requested data to enable us to ensure that the administrative record would be adequate to support GRASE determinations for the eligible sunscreen dosage forms. In particular, we emphasized that additional safety and efficacy data would be needed to support final monograph status for spray dosage forms. We also announced that we were issuing a draft guidance document (discussed above) explaining the Agency's intended enforcement policy for sunscreens marketed pursuant to the monograph system, including with respect to dosage forms. The Agency's approach to enforcement of spray sunscreens is now described in the 2018 Final Guidance. IV. Scope of This Rulemaking
Eligibility for inclusion in an OTC monograph was originally limited to active ingredients and other conditions that had been used in drugs marketed in the United States prior to the inception of the OTC Drug Review in 1972. After publication of the final sunscreen monograph in 1999, FDA published its TEA regulation (§ 330.14), (67 FR 3060 at 3074, January 23, 2002), which sets forth criteria and procedures by which OTC drugs initially marketed in the United States after the OTC Drug Review began and OTC drugs without any U.S. marketing experience can be considered for inclusion in the OTC drug monograph system. Congress later passed the SIA, which, among other things, supplements FDA's TEA regulations for OTC sunscreen drug products (21 U.S.C. 360fff through 360fff-7) (2014).
This proposed rule addresses the GRASE status (and conditions of use applicable to) sunscreen drug products containing active ingredients listed in the Stayed 1999 Final Monograph. It does not address the pending sunscreen active ingredients that were originally submitted under the procedures established in the TEA regulation and are now being addressed through the SIA process.[10] As discussed further in section VII, however, the safety data we described as necessary to evaluate the safety and effectiveness of sunscreen products containing those active ingredients are the same as what we are now describing as needed to establish that the active ingredients listed in the Stayed 1999 Final Monograph are GRASE for use in sunscreen products. We are not revisiting the contribution that the active ingredients listed in the Stayed 1999 Final Monograph make to the effectiveness of sunscreens. The Agency has not received information suggesting that it is necessary to revisit its prior decision about the effectiveness of the active ingredients at this time.
V. Legal Authority
We are issuing this proposed rule under sections 201, 301, 501, 502, 503, 505, 510, 586E, 701, 702, 703, 704, and 721 of the FD&C Act and under section 351 of the Public Health Service Act (42 U.S.C. 262).
VI. Need for Additional Safety Information
A. Increased Consumer Exposure to Sunscreen Active Ingredients
Consumer exposure to sunscreen active ingredients has increased dramatically since FDA began its initial safety evaluations of the sunscreen active ingredients at issue in this proposed rule. Many factors have influenced this increase, including the following:
Start Printed Page 6211- Significant increases in the number and types of consumers using sunscreen products (Refs. 2 and 3)
- Sunscreen products containing a greater number of active ingredients at greater concentrations (Ref. 4)
- Increased awareness of the risks of sun exposure and encouragement of routine sunscreen use by medical and public health authorities (see, e.g., Ref. 5)
- Evolving directions for use on sunscreen products instructing consumers to use greater amounts of sunscreen per application and to reapply sunscreen products more frequently (76 FR 35672 at 35678), codified as § 201.327)
- Expanding availability and use of many different types of sunscreen products, including daily-use products such as facial makeup, moisturizing creams, and lipstick
Relatively few sunscreen products were in use when the U.S. Army initially funded research into the development of effective sunscreen products for use by military personnel on aircraft carriers (and others routinely exposed to long periods of intense sunlight) during World War II (Ref. 2). The reach of sunscreen products began to broaden when they were later marketed for use specifically by consumers who sunburned readily (i.e., fair-skinned individuals) in situations of intentional sun exposure, such as sunbathing on a beach (Ref. 6). Sunscreen products are now routinely used by a much broader range of consumers for protection against many types of sun-induced skin damage, not just sunburn. Accumulating data demonstrate that increased sun exposure increases the risk of developing skin cancers and premature skin aging (Ref. 2). To help reduce the risk of these types of sun-induced skin damage, public health organizations (including FDA) have for years urged consumers to use sunscreen products along with other sun-protective behaviors like limiting time in the sun and wearing protective clothing (Refs. 7, 8, and 9).
Another factor driving increased consumer exposure to sunscreen active ingredients has been the introduction and widespread adoption of sunscreen products with higher labeled SPF values. The maximum SPF value proposed for sunscreen labeling has progressively increased from SPF 15 in the 1978 panel report, to SPF 30+ in the Stayed 1999 Final Monograph, to SPF 50+ in the 2011 Max SPF PR. To achieve these higher SPFs, many currently marketed products are formulated with more active ingredients combined together in higher concentrations than were generally combined in products when FDA's review of OTC sunscreens began. Increased knowledge about the role of UVA radiation in causing skin damage has also encouraged the formulation of broad spectrum products with combinations of active ingredients designed to achieve protection against both UVA and UVB radiation. In addition, other widely used products, such as facial makeup, moisturizing creams, and lipsticks, have had sunscreen active ingredients added to their formulations. These trends are reflected in the evolution of the current labeling provisions for sunscreen products regulated under the OTC monograph system.
Changes in the instructions for using these sunscreen products have also contributed to increased use of, and exposure to, sunscreen active ingredients. The labeling recommended by the advisory panel in 1978 simply instructed consumers to apply sunscreen products liberally and to reapply after swimming or excess perspiration (43 FR 38206 at 38215). The labeling currently required, by contrast, encourages consumers to always use a broad spectrum SPF 15 or higher product, to use sunscreen products regularly, and to apply them generously/liberally 15 minutes before sun exposure and at least every 2 hours or more frequently when swimming or sweating (§ 201.327(e)).
B. Emerging Safety Concerns
In recent years, a growing body of data has suggested that the transdermal absorption of some sunscreen active ingredients is greater than previously thought, and thus may raise previously unevaluated safety concerns, including the potential for reproductive, developmental, or carcinogenic effects. As discussed in further detail in section VIII.C.1.a, newly available information suggests, for example, that there is the potential for toxicity associated with the transdermal absorption and systemic availability of oxybenzone. This new information about absorption and potential safety risks is inadequate, by itself, to support an affirmative conclusion that products containing the active ingredients at issue are not safe. Coupled with the lack of clinical pharmacology and nonclinical safety data for certain sunscreen active ingredients, however, it leads us to conclude that, for some sunscreen active ingredients, the current record does not include adequate evidence of safety to satisfy the applicable legal standards for general recognition of safety and effectiveness as set forth in § 330.10.
VII. Framework for Evaluation of Safety Data
In light of these safety concerns, FDA held a meeting of its Nonprescription Drugs Advisory Committee (NDAC) on September 4 and 5, 2014, to discuss the scope of safety testing that should be conducted to support general recognition of safety and effectiveness for active ingredients for use in nonprescription sunscreen products. FDA proposed the following safety testing paradigm:
Clinical data:
- Dermal irritation and sensitization testing
- Phototoxicity and photoallergenicity testing
- Human maximal use bioavailability studies
- Postmarketing adverse event reports
Nonclinical (toxicology) data:
- Dermal carcinogenicity
- Systemic carcinogenicity
- Developmental and reproductive toxicity (DART)
- Toxicokinetics
- Additional testing when data suggest a concern about other long-term effects, such as endocrine effects
There was consensus among the committee members that FDA's proposed framework was a good starting point (Ref. 10). In November 2015, FDA published a draft guidance for industry, “Over-the-Counter Sunscreens: Safety and Effectiveness Data” (Draft Safety and Effectiveness Data Guidance) (see 80 FR 72975, November 23, 2015), which described and requested comment on the safety and effectiveness data necessary to determine whether an OTC sunscreen active ingredient or combination of active ingredients evaluated under the SIA was GRASE when used under specified conditions. FDA finalized this guidance in November 2016, after considering public comment on its draft recommendations (Ref. 11).[11] The recommendations in this guidance reflect FDA's scientific expertise, existing technical guidance, experience from reviewing safety and efficacy data submitted for GRASE review of sunscreen active ingredients under the OTC Drug Review, and input from and Start Printed Page 6212concurrence by outside scientific experts.
All sunscreens marketed without an NDA are subject to the same standard: General recognition of safety and effectiveness. Accordingly, as noted previously, the data that we expect to be necessary to evaluate the safety and effectiveness of the sunscreen monograph active ingredients are the same as those we recommended as necessary to evaluate the safety and effectiveness of sunscreen active ingredients previously considered under the procedures established in the TEA regulation and now being considered pursuant to the framework established by the SIA (see Safety and Effectiveness Data Guidance (Ref. 11)).
The studies described in this section are generally needed for FDA to determine that a sunscreen active ingredient is GRASE for use in nonprescription sunscreens. Specific data gaps for individual active ingredients depend on the quality and quantity of available safety data, and are identified in section VIII. As described in that section, those active ingredients for which the existing public record contains sufficient data to support a positive GRASE finding are proposed as Category I. Those for which additional data are necessary are proposed as Category III. In addition, in evaluating the existing safety data for the active ingredients listed in the Stayed 1999 Final Monograph, FDA determined that the risks associated with two of these ingredients outweigh their benefits. As discussed in further detail in section VIII.B, FDA is therefore proposing that these two ingredients are Category II because sunscreens containing these ingredients would not be GRASE.
A. General
FDA's OTC drug regulations identify the general types of safety information that should be submitted as evidence that an OTC drug is GRASE for use as labeled (§ 330.10(a)(2)) and the standard by which safety is to be judged (§ 330.10(a)(4)(i)). When applying these regulations to each drug, FDA uses its scientific expertise to determine what constitutes “adequate tests by methods reasonably applicable to show the drug is safe under the prescribed, recommended, or suggested conditions of use” (§ 330.10(a)(4)(i)).
FDA recognizes the contribution that broad spectrum sunscreens with an SPF value of 15 or higher can make to decreasing the risk of skin cancer and early skin aging caused by the sun if used as directed with other sun protection measures. To protect the public health, however, it is also important for FDA to balance the potential benefits of these sunscreen products to consumers against their potential risks. Providing an adequate safety margin for OTC sunscreen active ingredients and finished sunscreen products is a key element of FDA's risk assessment. A safety margin calculation takes the highest animal NOAEL and estimates a maximum safe level of exposure for humans. Because animal studies do not always predict effects in humans, the actual threshold for an effect in humans may be different (i.e., higher or lower) than in the species tested. The human sensitivity to a drug is often unknown. To account for this, the predicted safe exposure level in humans that is reflected in the safety margin is well below where toxicities were seen in animals.
In determining the specific testing and other data needed to adequately demonstrate that an OTC sunscreen active ingredient is safe, FDA considers both the circumstances under which OTC sunscreen products are intended to be used by consumers (i.e., the conditions of use) and current scientific knowledge and assessment technology. FDA's approach to the clinical safety evaluation of OTC sunscreen active ingredients is based on our current scientific understanding regarding safety evaluation of topical drug products for chronic use, and thus is generally consistent with the safety data requirements that would apply to an NDA for a chronic-use topical drug product (i.e., topical safety studies (irritation, sensitization, and photosafety); bioavailability (absorption); and evaluation of adverse events observed in clinical studies).[12] In addition, the evaluation of adverse events reported during the commercial marketing of sunscreen products containing the ingredient and other postmarketing safety information is also relevant to safety.
FDA's approach to the nonclinical safety evaluation of these active ingredients takes into account their lengthy marketing history in the United States. In contrast to nonclinical data requirements for a chronic-use topical drug product NDA, which include results from comprehensive nonclinical pharmacology and toxicology safety testing, the approach to nonclinical safety testing in this proposed rule is largely focused on potential long-term adverse effects or effects not otherwise readily detected from human use (i.e., carcinogenicity and reproductive toxicity). Additional testing beyond what is described below may be recommended for active ingredients for which data suggest a concern about other long-term effects, such as hormonal disruption.
In addition, although sunscreen products are typically formulated with two or more active ingredients, the framework described below contemplates that testing will be performed using formulations that include one active ingredient. Generally, unless data suggest that there may be a safety or efficacy concern with a particular combination of active ingredients, we anticipate that an active ingredient that is found to be GRASE for use in sunscreens could be combined with other active ingredients that are also GRASE for use in sunscreens. If data suggest that there may be a safety or efficacy concern with a particular combination of active ingredients (or active and inactive ingredients), additional data may be necessary to support a positive GRASE determination for sunscreens containing that combination.
The following sections describe the specific safety data that FDA expects the Agency will need to determine whether an active ingredient is GRASE for use in sunscreens.
B. Clinical Safety Testing
1. Human Dermal Safety Studies
Human dermal safety studies for topical products in which exposure to light after application is anticipated generally consist of two sets of studies—those conducted without specific exposure to light and those conducted to assess reactions after UV exposure (photosafety studies) (Ref. 12). The studies usually consist of dermal irritation patch testing, dermal sensitization patch testing, dermal phototoxicity testing, and dermal photoallergenicity testing.
Because marketed sunscreen products typically contain a combination of active ingredients, and product formulations frequently change, it is difficult to determine causal links between individual active ingredients and reported irritation and hypersensitivity adverse events associated with a particular product. Therefore, FDA generally expects to use data from human dermal irritation studies, human dermal sensitization studies, and human dermal photosafety studies, in conjunction with postmarketing adverse event data, to inform GRASE determinations and labeling. Nonetheless, in some cases, it may be reasonable to omit human Start Printed Page 6213dermal irritation studies, human dermal sensitization studies, and/or human dermal photosafety studies, depending on the rigor of available postmarketing safety information. For example, if FDA concludes that there is a positive risk-benefit profile for a sunscreen active ingredient, but that it is known to be a sensitizer, it may be possible to develop safety labeling to address this risk without data generated in the human dermal safety studies described below (see, e.g., section VIII.C.1.a).
a. Human dermal irritation and sensitization studies. Studies of dermal irritation and sensitization, using the repeat insult patch test or other relevant tests, are elements in the safety evaluation of topical drug products that, like sunscreens, are applied to the skin repeatedly over long periods of time. Designed to detect the potential for local dermatologic events with fewer subjects than might be observed in larger clinical trials, these tests often employ product application that is more frequent and/or for longer duration than proposed clinical dosing. In dermal irritation studies, a test substance is applied to a small pad (patch) and affixed to the test subject's skin, usually on the back, to determine whether the ingredient causes direct skin toxicity. Dermal sensitization studies are conducted similarly but are designed to detect immunologically mediated reactions, which require prior exposure to the allergen.
Nonprescription sunscreens regulated under the OTC monograph system may be used in many product formulations, including those yet unknown. Therefore, cumulative irritation studies that evaluate the sunscreen active ingredient at the highest concentration for which a GRASE determination is sought should be conducted using the ingredient in an appropriate vehicle, using the vehicle alone, and using both negative and positive controls. The evaluation should include scoring of erythema, edema, and a papular response or skin erosion.
Dermal sensitization studies, conducted to detect immunologically mediated reactions, should be conducted in three phases: (1) The induction phase (3 weekly applications for 3 weeks); (2) the rest phase (no product application for 10 to 14 days); and (3) the challenge phase (patch applications to new sites for 48 hours with a confirmatory rechallenge to exclude false positives).
Although FDA recommends separate dermal irritation and sensitization studies, it may be appropriate to combine irritation and sensitization studies in the same study as long as a sufficient number of subjects are included for sensitization evaluation.
b. Human photosafety studies. Topically applied dermatologic drug products should be tested for photosafety if they absorb light in the UVA, UVB, or visible spectra. Photosafety evaluations of sunscreen active ingredients that absorb light should consist of skin photoallergenicity and skin phototoxicity testing. Photoallergy is an immunologically mediated reaction to a chemical, initiated by the formation of photoproducts (e.g., protein adducts) following a photochemical reaction. Similar to dermal sensitivity testing described above, photoallergy tests use an induction/rest/challenge/rechallenge multiphase design to assess erythema, edema, and vesiculation. Phototoxicity (or photoirritation) is an acute light-induced tissue response to a photoreactive chemical. Phototoxicity testing typically includes a test patch, a vehicle patch, and a sham patch application for 24 hours, followed by UV light exposure of the test area. A second set of patch application areas not irradiated with light serves as a control. FDA expects that, to support a GRASE finding, photosafety studies of sunscreen active ingredients that absorb light will need to be conducted using the active ingredient at the highest concentration for which a GRASE determination is sought in an appropriate vehicle, using the vehicle alone, and with a negative control.
2. Human Absorption Studies/Maximal Usage Trial
Because nonprescription sunscreens are topically applied, a critical safety consideration is whether dermal application results in skin penetration and systemic exposure to their active ingredients and, if so, to what extent. This information helps identify potential safety concerns and helps determine whether an adequate safety margin exists within which an active ingredient is GRASE for use in sunscreens.
The principal barrier to topical drug product penetration is the multilayered, lipid-rich stratum corneum. The passage of any drug product through this layer is influenced by many factors, including the drug product's physicochemical features, molecular weight, and vehicle/formulation properties. Vehicle/formulation properties are particularly important because the choice of vehicle can markedly affect the permeation potential of a drug product. Effects can range from simple hydration of the stratum corneum by occlusive vehicles/formulations to direct permeation enhancement by solvent effects on the lipids in the stratum corneum. Products absorbed through the skin have the potential to cause systemic adverse effects, affecting the safety assessment. Because sunscreens are intended to work at the skin's surface, systemic absorption may also lower efficacy, affecting the efficacy assessment. Such considerations ultimately weigh into the risk-benefit calculus FDA uses to determine whether an OTC sunscreen containing a given active ingredient is GRASE.
Since the mid-1990s, topical product NDAs have included a Maximal Usage Trial (MUsT) as part of the clinical pharmacology/bioavailability assessment. A MUsT is designed to capture the effect of maximal use on absorption into the blood with standard pharmacokinetic assessments (e.g., Cmax, Tmax,[13] area under the curve, half-life, clearance, and volume of distribution) (for further information about conduct of a MUsT, see Ref. 13). For a topical product NDA, the MUsT is usually conducted in subjects with the disease of interest, where disrupted skin is a feature. In situations where disrupted skin is not a feature of the condition being treated or the topical product is intended for prevention of disease (e.g., sunscreens), the MUsT for a topical product NDA should be conducted in subjects with healthy, intact skin. The MUsT for a topical product NDA is conducted with the specific product formulation for which approval is sought applied at the upper limit of surface area involvement that is studied in the phase 3 clinical trials and is proposed for labeling. For example, if the proposed labeling of an acne product permits the product to be used on up to 30 percent of body surface area, that would be the coverage evaluated in the MUsT.
We expect that data from a MUsT will be needed to support an adequate assessment of safety for most sunscreen active ingredients (Ref. 10). Because sunscreen products regulated pursuant to the OTC monograph system may include active ingredients in a variety of formulations, FDA recommends that a MUsT be conducted under maximal use conditions employing a minimum of four formulations, containing the sunscreen active ingredient as the only active ingredient.[14] These formulations Start Printed Page 6214should be prepared using vehicle/formulation systems that are appropriate for sunscreen topical products (e.g., they are deployable and spreadable) that represent real-world marketed formulations, and that are expected to produce the highest in vivo absorption. Justification for the formulations chosen, including results of in vitro testing using a human cadaver skin permeation system (e.g., static cell, also known as vertical diffusion cell) (Refs. 14 and 15), should be included in the study protocol. The protocol should contain sufficient detail for others to reproduce the formulations and manufacturing process.[15]
FDA anticipates that the use of multiple formulations will help identify the overall absorption potential of the sunscreen active ingredient of interest. The MUsT should be conducted in subjects with healthy, intact skin [16] at the highest concentration of the ingredient for which a GRASE determination is sought. Based on recommended sunscreen use on all exposed skin, the exposed area should include at least 75 percent of the body surface area. Data from the formulation that produces the highest in vivo absorption would then be used to determine the safety margin.
The assay used in the MUsT should be properly validated according to current good laboratory practices (21 CFR part 58). Additionally, the Agency's most current guidance on bioanalytical method validation may be found by searching at https://www.fda.gov/RegulatoryInformation/Guidances/default.htm. The assay's limit of quantitation-limit of detection should be sufficiently low to allow a signal-to-noise ratio that ensures confidence in detection of a concentration of 0.5 nanogram (ng)/milliliter (mL) for the compound of interest in the receptor fluid.
An important consideration for designing a MUsT is that it should include testing for a duration that allows for the attainment of steady state levels to ensure that maximum penetration of the ingredient has taken place and to optimize the chances of the ingredient being detected. Thus, for sunscreen active ingredients, FDA expects that single application studies would be inadequate. Because the subjects in a MUsT represent an enriched dataset in the upper range of exposures, safety-related data (such as vital signs, adverse events) from the study's regularly scheduled physical examinations should also be collected. We strongly encourage consultation with FDA about MUsT protocols before beginning the trial.
Finally, as discussed further in section VIII.D, if the sunscreen active ingredient is determined to be GRASE for use in sunscreens, the sunscreen monograph, when finalized, must set out the conditions under which any future sunscreen containing that active ingredient will be GRASE and not misbranded. As such a condition, FDA is considering certain final formulation testing to address the potential for transdermal absorption and its impact on safety. FDA anticipates that the formulation that produces the highest in vivo absorption in the MUsT would be appropriate to designate as a standard control formulation for future in vitro human cadaver skin permeation system testing (e.g., a static or vertical diffusion cell) of each final sunscreen formulation that includes that active ingredient. If such testing were included as a condition in a final sunscreen monograph, and if in vitro permeation of the sunscreen active ingredient in the final product formulation were equal to or less than the value from in vitro testing of the standard control formulation (that was shown by the MUsT to have the highest degree of systemic absorption), FDA anticipates that the safety margin previously calculated would be considered adequate to support the safety of the finished formulation.
3. Pediatric Considerations
Young children have a larger ratio of skin surface to body volume than adults, which can increase a child's systemic exposure to topically applied drug products. In addition, growing children have greater potential to experience deleterious developmental effects from drug exposure. If the calculated safety margin for an active ingredient (based on nonclinical results and human MUsT) is relatively small, FDA will exercise its scientific judgment to determine whether a sunscreen active ingredient MUsT in young children or other studies are warranted to ensure that the safety margin for marketed products containing the ingredient is within an acceptable range for this population.
C. Nonclinical Safety Testing
1. Carcinogenicity Studies: Dermal and Systemic
FDA generally recommends carcinogenicity studies for any pharmaceutical with an expected clinical use (either intermittent or continuous) of at least 6 months (Ref. 17). The animal carcinogenicity studies help characterize the potential tumor risks associated with use of a sunscreen active ingredient in human beings by identifying any observed tumors by type, the level of exposure at which tumors occur, and the highest level of exposure at which no adverse effects occur, referred to as the NOAEL. As noted earlier, FDA intends to use the NOAEL in determining the safety margin for human exposure to sunscreens containing the active ingredient. In addition to detecting carcinogenic potential, carcinogenicity studies in animals can also help to identify other systemic or organ toxicities that may be associated with the sunscreen active ingredient.
FDA expects that a dermal carcinogenicity study involving application of the test article to the skin of mice or rats for 2 years will thus need to be conducted to support a GRASE finding for the active ingredient unless the ingredient has been demonstrated not to reach the viable layers of the skin where it could impact skin tumor development. FDA also considers it important to study the effects of systemic exposure if human bioavailability data show that dermal application of a particular formulation results in skin penetration and systemic exposure to the active ingredient. Therefore, we expect that a second carcinogenicity study by a route that produces systemic exposure will also be needed to support the safety of a sunscreen active ingredient, if systemic exposure is observed in the bioavailability data. This can be a 2-year study or a shorter (usually 6 months) alternative carcinogenicity model, and it should be conducted in a species different from that used in the dermal carcinogenicity study. FDA notes that the absence of a carcinogenicity signal from an alternative transgenic carcinogenicity study (e.g., TgRasH2 mouse) would likely support the safety of a sunscreen active ingredient. If a carcinogenicity signal were observed in such a study, however, the study could not be used to support the safety of a sunscreen active ingredient because there would be no basis for calculating Start Printed Page 6215a safety margin with this study (Ref. 18). All carcinogenicity studies, regardless of route, should assess a full panel of tissues.[17]
FDA expects that a systemic carcinogenicity study would not be needed to support a GRASE determination for a sunscreen active ingredient if an adequately conducted human pharmacokinetic MUsT resulted in a steady state blood level less than 0.5 ng/mL, and an adequately conducted toxicology program did not reveal any other safety signals for the ingredient or any known structurally similar compound indicating the potential for adverse effects at lower levels. The threshold value of 0.5 ng/mL is based on the assessment that the level would approximate the highest plasma level below which the carcinogenic risk of any unknown compound would be less than 1 in 100,000 after a single dose. This threshold value is consistent with the Threshold of Toxicological Concern concept, which was applied to impurities in the ICH guidance for industry “M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk” (Ref. 19). FDA expects that the 0.5 ng/mL concentration will be sufficiently above the assay's limit of quantitation—limit of detection to allow a signal-to-noise ratio that ensures confidence in either the derived concentrations (in the case of “exaggerated” values) or lack of concentrations.
2. Developmental and Reproductive Toxicity Studies
FDA expects that DART studies will need to be conducted to evaluate the potential effects that exposure to the sunscreen active ingredient may have on developing offspring throughout gestation and postnatally until sexual maturation, as well as on the reproductive competence of sexually mature male and female animals (Ref. 20). As with systemic carcinogenicity studies, we expect that studies to assess fertility and early embryonic development, and pre- or postnatal toxicity in rats will not be needed if an adequately conducted human MUsT shows a steady state blood level less than 0.5 ng/mL, and an adequately conducted toxicology program produces no signals indicating that the ingredient (including its clinically relevant metabolites) or any known structurally similar compound interacts with related pathways.[18] We expect that effects on embryofetal development will need to be assessed in rats and rabbits in all cases.
Gestational and neonatal stages of development may be particularly sensitive to active ingredients with hormonal activity (endocrine disruption). For this reason, these studies should include assessments of endpoints such as vaginal patency, preputial separation, anogenital distance, and nipple retention, which can be incorporated into traditional DART study designs to assess potential hormonal effects on the developing offspring. Behavioral assessments (e.g., mating behavior) of offspring, which may detect neuroendocrine effects, should also be performed (Ref. 21).
3. Toxicokinetics (Ref. 22)
Animal toxicokinetic data should also be collected for sunscreen active ingredients, as these data provide an important bridge between toxic levels seen in animal studies and any potential human adverse events associated with systemic exposure to the sunscreen's active ingredient. Toxicokinetic measurements are usually obtained during the course of ongoing nonclinical toxicity studies, such as carcinogenicity or DART studies, rather than through separate studies.
D. Postmarketing Safety Data
In addition to the active ingredient safety data already described, FDA's GRASE evaluation also takes into consideration publicly available information about serious adverse drug experiences and known or expected adverse effects associated with commercially marketed products that contain the active ingredient(s) under consideration.
E. Sunscreens Containing Nanomaterials
We note that FDA is not proposing to categorically classify sunscreen products manufactured using nanotechnology (or containing nanomaterials) as GRASE or not GRASE solely based on this characteristic. Nanotechnology is used to create, explore, or manipulate materials measured in nanometers (nm) (billionths of a meter), and has applications in a wide range of products, including OTC sunscreens. Such materials generally have dimensions between approximately 1 and 100 nm (Ref. 23). Materials at such small sizes can have different chemical or physical properties or biological effects compared to larger-scale counterparts, making possible a variety of functional effects, and also potentially affecting the safety, effectiveness, or regulatory status of FDA-regulated products.
FDA has not established regulatory definitions of nanotechnology, nanomaterial, nanoscale, or other related terms. As described in FDA's guidance for industry “Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology” (Nanotechnology Considerations Guidance) (Ref. 24), at this time, when considering whether an FDA-regulated product involves the application of nanotechnology, FDA asks
(1) Whether a material or end product is engineered to have at least one external dimension, or an internal or surface structure, in the nanoscale range (approximately 1 nm to 100 nm).
In addition, because materials or end products can also exhibit related properties or phenomena attributable to a dimension(s) outside the nanoscale range of approximately 1 nm to 100 nm that are relevant to evaluations of safety, effectiveness, performance, quality, public health impact, or regulatory status of products, we will also ask:
(2) Whether a material or end-product is engineered to exhibit properties or phenomena, including physical or chemical properties or biological effects, that are attributable to its dimension(s), even if these dimensions fall outside the nanoscale range, up to 1 micrometer (µm) (1,000 nm).
We will apply these considerations broadly to all FDA-regulated products, including sunscreen products. For the purpose of this proposed rule, we use the term “nanomaterial” generally to refer to materials falling within either point 1 or 2 above. The use of this term in this manner is consistent with its use in FDA's nanotechnology-related guidances, including FDA's Nanotechnology Considerations Guidance.
Nanomaterial forms of the active ingredients zinc oxide and titanium dioxide have been used in marketed OTC sunscreens. In addition to nanomaterial forms of zinc oxide and titanium dioxide, other nanomaterials are also reported to have been used, or promoted or studied for possible use, in sunscreen products (Ref. 25).Start Printed Page 6216
As discussed in further detail in section VIII.A, having examined the scientific information in the record, including for nanomaterial forms of zinc oxide and titanium dioxide, FDA is not now proposing conditions of use for these two active ingredients under the sunscreen monograph that distinguish nanomaterials from other forms of these ingredients. As indicated above, FDA also does not propose to categorically classify sunscreen products that are manufactured using nanotechnology or contain nanomaterials as GRASE or not, solely on that basis. Manufacturers of products containing nanomaterials marketed under the OTC sunscreen monograph remain responsible for ensuring that the product satisfies all applicable legal requirements. FDA encourages manufacturers of such products to consult with FDA to facilitate a mutual understanding of specific scientific or regulatory issues relevant to their product.
FDA invites comment on the following topics:
- Specific nanomaterials or types of nanomaterials that have been used or proposed for use in OTC sunscreen products
- Concerns about sunscreen product safety, effectiveness, or quality associated with the use of nanomaterials in OTC sunscreen products, with supporting data
- Need for, and proposals of, specifications or limitations for particular nanomaterials for use in OTC sunscreen products
- Any particular nanomaterials that you believe should not be permitted for use in OTC sunscreen products, along with supporting scientific information
- FDA's proposed regulatory approach and/or alternative regulatory approaches to the use of nanomaterials in OTC sunscreen products
VIII. Existing Safety Data for Sunscreen Active Ingredients
In the remainder of this section, we discuss the existing data and data gaps for each of the sunscreen monograph active ingredients and explain why we propose that these active ingredients are GRASE or not GRASE for use in sunscreens. Those ingredients for which the existing data are sufficient to support a positive GRASE determination are proposed as Category I. Those ingredients for which additional data are necessary before a GRASE determination can be made are proposed as Category III. In cases where FDA's evaluation of the existing safety data caused us to determine that the risks associated with the ingredients outweigh their benefits, the ingredients are proposed as Category II.
A. Ingredients Proposed as Category I
Based on our review of the publicly available data for these ingredients, both zinc oxide and titanium dioxide are proposed as Category I.
1. Zinc Oxide
Our review of the scientific literature, submissions to the sunscreen monograph docket, and adverse event reports submitted to FAERS has produced sufficient safety data on zinc oxide to support a proposal that a sunscreen containing up to 25 percent zinc oxide would be GRASE under the conditions proposed in this rulemaking and the general conditions required in part 330. This proposal is based in significant part on the existing, substantial evidence that zinc oxide (including particles on the nanoscale, i.e., approximately 1 to 100 nm) does not penetrate into or through human skin to any great extent and, to the extent any de minimis penetration occurs, does not result in adverse health effects, given the high levels of endogenous zinc in the human system.
a. Background. Zinc oxide is an inorganic, mineral compound. Because of its ability to reflect UVA wavelengths, zinc oxide is frequently used in sunscreens to help establish broad spectrum protection (Ref. 26). While larger particles of zinc oxide used in sunscreens (greater than approximately 100 nm) may impart an opaque, white color to the product, zinc oxide is also manufactured in smaller particle sizes (less than approximately 100 nm) to reduce this white/opaque appearance (Refs. 27 and 28). In addition to its use in sunscreens, zinc oxide is also used in non-sunscreen ointments, pastes, and lotions for various skin disorders because of its protective, astringent, and antiseptic properties (Ref. 29).
b. Discussion. Zinc oxide is insoluble in water and largely insoluble in biological fluids.[19] This insolubility precludes the possibility of its systemic absorption from topical application of sunscreen products beyond a de minimis amount, even if zinc oxide is included at its maximum eligible concentration of 25 percent and regardless of the formulation of the product. The available studies on the dermal penetration of zinc oxide, further discussed below, confirm that its penetration—regardless of particle size—is primarily limited to the upper layers of the non-living stratum corneum, with most penetration occurring only into skin folds and furrows or hair follicles. These studies show that zinc oxide particles do not penetrate down into the viable dermis to any significant extent. Any de minimis transdermal penetration that may occur does not result in adverse health effects, because the tiny amount of zinc oxide particles that achieve transdermal absorption, if any, would dissociate into zinc and oxygen ions, both of which are naturally occurring elements in the human body (Ref. 30). Zinc is the 14th most common element in the human body and is essential for all living things; the average human body contains about 2.0 to 2.5 grams of zinc, and normal dietary intake of zinc is about 15 milligram (mg) per day (Refs. 30 and 31). Homeostatic mechanisms in the body regulate zinc's absorption, distribution, cellular uptake, and excretion (Ref. 31). Similarly, any oxygen absorbed through the skin is nonharmful, as oxygen is plentiful in the human body and essential for life.
Our search of the literature on zinc oxide revealed four recent studies about zinc oxide's penetration into human skin, which confirm our expectations based on the physical properties of this compound. The first two studies (conducted by Leite-Silva et al. and Darvin et al.) examined the penetration of zinc oxide into the skin using multiphoton tomography (Refs. 32 and 33). Both studies showed a lack of overall permeation of zinc oxide beyond a few cell layers, except in the case of furrows and wrinkles (Refs. 32 and 33). The second two studies—a pilot and subsequent full trial conducted by Gulson et al.—evaluated the penetration of nanoscale zinc oxide into the skin and the bloodstream using a stable isotope tracing method (Refs. 34 and 35). Although the Gulson studies found that a minimal amount of topically applied zinc was absorbed, the absorption observed was at levels that are orders of magnitude less than daily nutritional intake and well below what would be of concern for a naturally occurring element in the body subject to homeostatic mechanisms (Ref. 36). An additional porcine study found (as discussed in our 2012 response to a citizen petition submitted by the International Center for Technology Assessment and others (Docket No. FDA-2006-P-0213-0003) (ICTA Petition Response)), that although sunburn caused by UVB rays increased the penetration of zinc oxide into the Start Printed Page 6217non-living stratum corneum, there remained minimal penetration of zinc oxide into the epidermal and dermal layers of the skin (Ref. 37). Because topically applied zinc oxide particles do not enter systemic circulation to any meaningful extent, we do not consider a MUsT to be necessary to support the safety of this ingredient.
In addition to the studies described above, we also located two studies evaluating the clinical safety of topically applied zinc oxide in which zinc oxide (25 percent) was used as a medicated occlusive dressing on the lower arms of healthy volunteers (Refs. 38 and 39). In these studies (which were designed to maximize potential absorption and identify any resulting adverse events), even with the increased dermal or epidermal zinc levels resulting from occlusion, there still were no adverse skin events. Our review of the available human dermal safety studies on zinc oxide [20] also identified data showing that test material containing up to 25 percent zinc oxide did not induce human irritant, photoirritant, allergic, or photoallergic reactions. No human pathological phototoxicity or significant human photosensitization reaction indicative of skin irritation were noted either. The literature supporting the safety of skin protectant drug products containing zinc oxide [21] reinforce these clinical safety findings. Our review in this area is also consistent with the conclusion of the European Commission's Scientific Committee on Consumer Safety that the use of nanoscale zinc oxide in sunscreens at a concentration of up to 25 percent does not pose a risk of adverse effects in humans after topical application (Ref. 40).
A very small number of rash and hypersensitivity reports for sunscreens containing zinc oxide were located in FAERS. With a single exception, the sunscreens involved contained two or more active ingredients, making it difficult to attribute causation to a specific active ingredient. Unlike other sunscreen ingredients with a known hypersensitivity risk, we did not identify any reports in FAERS or in the literature with features suggestive of a causative link, such as skin test results positive for zinc oxide. In addition, there is an extremely large safety database of zinc oxide use in other topical products, including for the treatment of diaper rash in infants. This corroborates the negative results in human studies for irritation, photoirritation, allergy, and photoallergy that support our proposed finding regarding the safety of sunscreens containing this ingredient under the conditions proposed. Reports of non-hypersensitivity-related clinical safety issues with zinc oxide were infrequent and not serious. For these reasons, we do not consider additional clinical studies (including photosafety, irritation, or sensitization studies) to be necessary for this ingredient.
Dermal carcinogenicity studies have not been conducted for zinc oxide. In general, as discussed in section VII.C.1, adequate tests for safety of an active ingredient for use in topical products for chronic use (such as a sunscreen) would need to include dermal carcinogenicity studies if the active ingredient reaches the viable layers of skin where it could have a biological effect. Given the minimal penetration of zinc oxide below the non-living stratum corneum, there is no plausible mechanism by which zinc oxide could have an effect on skin tumor development. We are therefore proposing to find that zinc oxide is GRASE for use in sunscreens despite the lack of dermal carcinogenicity studies studying this ingredient.
Based on the minimal systemic exposure resulting from dermally applied zinc oxide, in particular when compared to endogenous zinc levels, we see no need for further nonclinical studies to support the safety of sunscreens containing zinc oxide, including systemic carcinogenicity studies, developmental and reproductive toxicity studies, or toxicokinetic studies.[22]
c. Conclusion. Our review of the available data from both animal and human studies and data on physical properties such as solubility leads us to conclude that the transdermal absorption of zinc oxide—regardless of particle size—from any topically applied sunscreen formulation is extremely unlikely, and that any de minimis absorption that may occur would not result in adverse health effects, given the high levels of endogenous zinc. The very low likelihood of any systemic absorption of zinc oxide in turn indicates that the safety margin for zinc oxide is large; accordingly, consistent with our approach to pediatric studies discussed in section VII.B.5, we do not consider pediatric studies to be needed for this ingredient. We propose to find that the currently available safety data provide sufficient evidence to demonstrate the minimal absorption, low dermal irritation, low allergenic sensitization and photoallergenicity, and low phototoxic potential of zinc oxide—regardless of particle size—up to 25 percent, and that these data support a finding that zinc oxide up to 25 percent is GRASE for use in sunscreens under the proposed conditions. Accordingly, we propose that zinc oxide is a Category I active ingredient.
2. Titanium Dioxide
For similar reasons, we propose that titanium dioxide is also a Category I active ingredient. Our review of information publicly available in the scientific literature, submissions to the sunscreen monograph docket, and FAERS has produced sufficient information to support a proposal that a sunscreen product containing up to 25 percent titanium dioxide would be GRASE under the conditions proposed in this rulemaking and the general conditions required in part 330.
a. Background. Titanium dioxide is an inorganic mineral compound consisting of small, crystalline-structured or amorphous particles. It is widely used as an excipient and is currently listed as an inactive ingredient in more than 60 approved drug products (including topical, oral, and inhalation products, among others) (Ref. 46). Titanium dioxide particles can be manufactured to have a variety of different dimensions, shapes (such as spheres or rods), and crystal polymorphs (such as anatase or rutile). Titanium dioxide (typically with particle dimensions ranging from 200 to 300 nm) is manufactured as a white powder for use as a white color pigment in pharmaceuticals. Manufacturers have also introduced processes that produce Start Printed Page 6218titanium dioxide with particle dimensions ranging from 15 to 50 nm to reduce its white/opaque appearance. Titanium dioxide particles used in sunscreens are also now often treated with chemical coatings (such as silicones, metal oxides, or organic acids) that are bonded to the exterior surface of the particles to, among other things, improve the aesthetic characteristics of the final formulation.
b. Discussion. Titanium dioxide is essentially insoluble in water and in biologic fluids (Ref. 47). As with zinc oxide, this lack of solubility prevents the transdermal absorption of more than a de minimis amount of titanium dioxide, regardless of either the concentration of titanium dioxide or the formulation of the product (Refs. 48 and 49). Further, unlike zinc oxide, which, if dissolved, would dissociate into zinc and oxygen (Ref. 50), the chemical stability of titanium dioxide is such that it does not dissociate under the conditions that exist in (or on) the human body (Ref. 51). Even if titanium dioxide were to dissociate into titanium and oxygen, titanium is unreactive in physiologic conditions, and (for this, among other, reasons) is frequently used in medical devices and structures implanted in the human body (Refs. 51 and 52).
The available studies on the transdermal absorption of titanium dioxide confirm that the skin is an effective barrier to the penetration of titanium dioxide, regardless of particle size—including those on the nanoscale (Refs. 53, 54, and 55). In our 2012 response to the ICTA Petition mentioned earlier, we described the then available information about the absorption of titanium dioxide nanomaterials and concluded that the “currently available literature indicates that insoluble nanomaterials of titanium dioxide used in sunscreens do not penetrate into or through human skin to produce adverse health effects when applied topically” (ICTA Petition Response at 26). Since that time, our search of the available literature has not revealed anything that would change this conclusion. Because topically applied titanium dioxide particles do not enter systemic circulation to any meaningful extent, we do not consider a MUsT to be necessary for this ingredient.
Given the lack of transdermal absorption of titanium dioxide beyond a de minims amount and, as a result, the very low likelihood of any systemic effects, we also do not consider additional nonclinical studies (including systemic carcinogenicity, developmental and reproductive toxicity, or toxicokinetic) to be necessary to support the safety of this ingredient.[23] Because titanium dioxide penetration beyond the non-living stratum corneum and into the viable layers of the skin is also minimal, as with zinc oxide, we do not consider dermal carcinogenicity studies to be needed for titanium dioxide either.
The inability of more than an extremely minimal amount of titanium dioxide to reach viable tissues that could have an immunologic reaction also prevents dermal irritation, sensitization reactions, and photosafety issues for this ingredient. Our search of the available literature on titanium dioxide identified nonclinical data reinforcing this, showing that dermal toxicity after dermal application of titanium dioxide in rodents is minimal (Refs. 57 to 60). Accordingly, we do not consider additional clinical photosafety, irritation, or sensitization studies to be necessary to support the safety of this ingredient. We note that the available studies on titanium dioxide evaluate products with titanium dioxide concentrations up to 10 percent. Given that the physical properties of titanium dioxide both preclude its penetration into or through the human skin regardless of concentration and make it unlikely that there would be dermal photosafety, irritation, or sensitization associated with titanium dioxide exposure (and that there is no data to suggest such photosafety, irritation, or sensitization would exist at higher concentrations), we propose that titanium dioxide—regardless of particle size—is GRASE for use in sunscreens at concentrations up to 25 percent, consistent with the level set in the Stayed 1999 Final Monograph.
In evaluating whether titanium dioxide is GRASE for use in sunscreen products, we have considered published literature indicating that nanoscale titanium dioxide can exhibit photocatalytic properties (Ref. 61). The literature indicates that the crystalline structure of titanium dioxide particles plays a role in this photocatalytic activity, and that the anatase crystalline polymorph is associated with greater photocatalytic activity than the rutile polymorph (Ref. 61). The European Commission has established limitations on the percentage of anatase crystalline polymorph in titanium dioxide to minimize photocatalytic activity.[24] Coating titanium dioxide particles has also been shown to minimize photocatalytic activity (and to limit particle clumping, which can have an impact on how products blend).[25]
In theory, if photocatalytic activity occurred when sunscreen products containing nanoscale titanium dioxide were exposed to light, it could result in the breakdown of other sunscreen active ingredients in these products. We have no evidence, however, that this in fact occurs in sunscreen products containing titanium dioxide or that there are any other negative impacts resulting from such photocatalytic activity. Accordingly, its potential for photocatalytic activity does not undermine our conclusion that titanium dioxide is GRASE for use in sunscreen products. Nonetheless, we invite comment (including supporting data) on whether sunscreens containing titanium dioxide are negatively impacted by the potential photocatalytic effects of that ingredient and, if so, to what extent; and on additional regulatory conditions, if any, that are necessary to address this potential issue.
We note, as well, that it is the responsibility of manufacturers to ensure that any inactive ingredients used in a drug product marketed pursuant to the OTC Drug Review, including coatings used to address photocatalytic activity or for other purposes, are safe and suitable for their intended use (see § 330.1(e)). FDA encourages manufacturers to contact the Agency regarding any specific coatings that they are considering for use in a topical sunscreen.
c. Conclusion. Given the chemical properties of titanium dioxide as insoluble and unreactive under physiologic conditions and the available studies showing that titanium dioxide does not penetrate into the skin or enter into systemic circulation to any meaningful extent, we consider the available safety data adequate to support a proposal that titanium dioxide is GRASE for use in sunscreens. As with zinc oxide, our proposal rests in significant part on the data showing that absorption of titanium dioxide into or through the skin is very unlikely and Start Printed Page 6219that any de minimis absorption that could theoretically occur would not result in adverse health effects. As a result, the safety margin here is large, and consistent with our approach to pediatric studies discussed in section VII.B.5, we therefore consider pediatric studies to be unnecessary for this ingredient.
B. Ingredients Proposed as Category II
FDA's review of the available safety data for PABA and trolamine salicylate have caused us to conclude that the risks associated with use of these ingredients in sunscreen products outweigh their benefits. Accordingly, we are proposing that these two ingredients are Category II.
1. Para-Aminobenzoic Acid
PABA use has decreased significantly in recent years because of, among other things, its adverse effects on skin and its discoloring and staining effect on clothing. Our review of more than 700 sunscreen brands sold in the United States (Ref. 63) indicates that PABA is in fact no longer being marketed in the United States.
A search of the scientific literature, submissions to the sunscreen monograph docket, drug approval documents from FDA and the European Medicines Agency, adverse event reports submitted to FAERS, and FDA Advisory Committee meeting reports (among other sources) has produced clinical safety data on PABA that supports a conclusion that a sunscreen containing PABA would not be GRASE. The available clinical information includes significant numbers of reports of allergic and photoallergic skin reactions to PABA, with rates of PABA-induced skin reactions potentially 8 percent or higher (Refs. 64 to 67). An 8 percent incidence is a serious concern: By comparison, only 34 hypersensitivity reactions associated with sunscreen products have been identified in FAERS since 1969.[26]
Further, PABA has the ability to cause cross-sensitization to structurally similar aromatic amines and nitro compounds (i.e., it can cause individuals exposed to it to develop sensitivity reactions to similar compounds) (Ref. 69). The list of compounds at issue includes a variety of widely used products, such as sulfonamide antibiotics (commonly used to treat a variety of infections, from urinary tract infections to certain types of pneumonia), thiazide diuretics (the number one recommended treatment for hypertension for certain communities), certain local anesthetics (such as benzocaine and procaine), and dyes (including para-phenylenediamine (a hair dye) and aniline dyes (used in medical products)) (Refs. 70, 71, and 72). Cross-sensitization to these products is a serious concern, as widespread PABA use could result in a significant increase in cross-reactivity with these agents and the incidence of allergic and photoallergic dermatitis, some of which are likely to be severe.
These safety issues alone are reason enough to find PABA not GRASE for use in sunscreens. In addition, however, data obtained from the urine samples of human subjects receiving topical PABA application shows that PABA also penetrates the skin and enters systemic circulation (Ref. 73). Because full MUsT studies for PABA have not been done, it is unclear to what degree such transdermal absorption takes place. However, one article in the published literature suggests that there is an association between autoimmune disorder and PABA use (Ref. 71), and we found one report each of hepatotoxicity (Ref. 74) and chronic interstitial nephritis (Ref. 75) after oral PABA administration. Although it is difficult to determine causality on the basis of such single reports, if a MUsT were to show absorption of PABA, these reports could represent an additional safety concern.
In addition, genotoxicity findings with PABA use have been largely negative in the absence of UV irradiation. Adequate assessments of the dermal carcinogenicity potential of PABA are unavailable, as are DART studies. If a MUsT were to show absorption of PABA, therefore, necessary studies would include dermal and systemic carcinogenicity studies, DART studies, and toxicokinetic studies. However, given that the above-described safety concerns associated with PABA are significant enough to place PABA in Category II, conducting such testing is neither appropriate nor ethical. We propose that PABA is not GRASE for use in sunscreens.
2. Trolamine Salicylate
We also propose that trolamine salicylate is not GRASE for use in sunscreens, and is, like PABA, a Category II active ingredient. As described in further detail below, there are significant safety concerns associated with the use of trolamine salicylate in sunscreen products. We propose that these concerns are sufficient to support a conclusion that a sunscreen containing trolamine salicylate would not be GRASE. We note that, as with PABA, our review of more than 700 sunscreen brands sold in the United States suggests that trolamine salicylate is no longer being marketed in sunscreens sold in the United States (Ref. 63).
a. Background. Trolamine salicylate is comprised of trolamine and salicylic acid. Salicylic acid is a non-steroidal anti-inflammatory drug (NSAID); it is the active moiety in aspirin, and has been widely used as an analgesic (i.e., pain relieving), anti-pyretic (i.e., fever reducing), and anti-inflammatory agent. In addition to these properties, salicylic acid inhibits platelet aggregation, which in turn inhibits blood coagulation. For this reason, some salicylic acid-containing products (such as aspirin) are used by consumers to help reduce cardiovascular adverse events, including myocardial infarction, stent thrombosis, and transient ischemic attacks.
Trolamine salicylate was included in the Stayed 1999 Final Monograph for sunscreens at a concentration of up to 12 percent. It was also proposed as a Category III active ingredient in the tentative final monograph for OTC external analgesic drug products (External Analgesic TFM) (“External Analgesic Drug Products for Over-the-Counter Human Use; Tentative Final Monograph,” 48 FR 5852 at 5855 (February 8, 1983)). The mechanisms of action for trolamine salicylate for these two drug categories are very different; to be effective as an external analgesic, trolamine salicylate must penetrate the skin and reach the relevant sites of action. The available evidence clearly establishes that trolamine salicylate is transdermally absorbed (Refs. 76 and 77). To be effective as a sunscreen, however, trolamine salicylate must be present on the surface of the skin so that it can reflect, scatter, or absorb UV radiation.
The directions for use for the two product categories differ significantly as well. The current requirements for sunscreen labeling include directions that the product should be applied to all skin exposed to the sun, that it should be used “regularly” to decrease the risk of skin cancer and early skin aging,[27] and that it should be reapplied at least every 2 hours (21 CFR 201.327). In contrast, currently marketed external analgesic products containing trolamine salicylate include directions for use stating that they should be applied to “affected areas,” that they should be reapplied no more than three to four Start Printed Page 6220times a day, and that use should be discontinued after 7 days.[28]
b. Significant safety concerns associated with use of trolamine salicylate as a sunscreen. FDA is concerned that use of trolamine salicylate as an active ingredient in sunscreens could cause serious detrimental health effects due to the anti-coagulation effects of salicylic acid. FDA located two case reports of serious coagulation-related adverse events associated with liberal dermal application of trolamine salicylate. The first case involved a surgical patient who experienced coagulopathy (impairment of the blood's ability to coagulate) at surgical sites in connection with use of topical trolamine salicylate (Ref. 76). Although the patient discontinued aspirin use 2 weeks before surgery per her doctor's instructions, she was unaware that use of a topical cream containing trolamine salicylate should have been stopped as well, and continued liberal application of the product to her knees for arthritis pain in the period leading up to her surgery. Four hours after surgery, the patient returned to the operating room bleeding profusely from all surfaces that had been operated on and experiencing massive bilateral hematomas. She lost more than 900 mL of blood.
In the second case, a patient taking warfarin (an anticoagulant) for atrial fibrillation and stroke prevention experienced a considerable increase in prothrombin time (i.e., the time it takes for blood to coagulate) after liberal application of trolamine salicylate to his neck and shoulders for pain relief (Ref. 78). The patient's prothrombin time had previously been in the therapeutic range of 1.3 to 1.5 times the control, but increased to 2.5 times the control during trolamine salicylate use. When trolamine salicylate use was discontinued, the patient's prothrombin time returned to 1.3 times the control.
FDA is also concerned that sunscreens containing trolamine salicylate could lead to other adverse effects associated with salicylic acid exposure. These include gastrointestinal distress and hemorrhage, ototoxic effects (i.e., impacts on hearing), hypersensitivity reactions, asthma exacerbations, acid-base imbalance, salt and water retention, liver injury, and Reye's Syndrome (in children). At high doses, acute salicylate toxicity (salicylism) may occur. Early symptoms of salicylism include tinnitus, vertigo, nausea, vomiting, and diarrhea; subsequent symptoms suggesting a more severe intoxication include altered mental status (ranging from agitation to lethargy), hyperpyrexia, noncardiac pulmonary edema, and coma.[29]
If trolamine salicylate were to be applied to all skin exposed to the sun and reapplied every 2 hours as directed in sunscreen labeling, the potential for transdermal absorption and systemic availability of substantial amounts of salicylic acid raises significant concerns about the potential for increased occurrence of the above-described adverse events. This is a particular concern given the widespread use of other OTC NSAID products with anti-inflammatory, analgesic, or anti-pyretic effects, which, combined with the use of sunscreens containing trolamine salicylate, may raise the anti-platelet effects experienced by consumers to problematic levels. Concerns relating to transdermal absorption may be especially acute for children, who have a higher surface-area-to-body-weight ratio than adults. FDA proposes that the above-described safety concerns are enough, by themselves, to support a finding that trolamine salicylate is not GRASE for use in sunscreens, and therefore, is a Category II active ingredient.
c. Data gaps. In addition, there are several categories of data about trolamine salicylate that FDA expects would be necessary to support a positive GRASE determination for its use in sunscreen products that are currently missing from the public record. For example, there is insufficient clinical dermal sensitization, irritation, and photosafety data for trolamine salicylate. Although the transdermal absorption of trolamine salicylate is well established, the record currently lacks a MUsT that would allow us to evaluate the extent of exposure to this ingredient when it is used as a sunscreen. Such data is important because it would allow FDA to interpret systemic toxicity findings in animal toxicology studies in the context of the amount likely to be absorbed from sunscreen use. Given the FDA recommendation that a MUsT for sunscreen use include application to a majority (75 percent at a minimum) of the body surface of each test subject, the above described safety concerns (including the potential for salicylism associated with exposure to high doses of trolamine salicylate) would raise significant ethical concerns about the conduct of a MUsT in these circumstances. Were it possible to ethically conduct a MUsT for this ingredient, and if such a MUsT showed significant transdermal absorption of trolamine salicylate, this would raise questions about whether enough of this ingredient remains present on the surface of the skin for it to function effectively as a sunscreen. As we noted in section VII.B.4, such considerations ultimately weigh into the risk-benefit calculus FDA uses to determine whether an active ingredient would be GRASE for use in sunscreens.
Although we have data addressing the toxicology profile of salicylate, adequately detailed nonclinical DART studies for trolamine and toxicokinetic data to interpret DART studies were also not found in the public record. Adequate DART information, if it were available, might reveal additional data needs (for example, to address any potential hormonal effects that may be identified). Dermal carcinogenicity data are available from the National Toxicology Program for trolamine in acetone and trolamine alone (applied neat).[30] In the absence of toxicokinetic data to interpret existing carcinogenicity studies, we cannot determine how the exposure in the animal studies relates to human exposure to trolamine from the use of trolamine salicylate as a sunscreen active ingredient.
d. Conclusion. For the reasons described above, FDA proposes that trolamine salicylate is not GRASE for use in sunscreens. The safety concerns associated with the use of trolamine salicylate as an active ingredient in sunscreens are significant enough to support classification of trolamine salicylate as a Category II ingredient. In particular, the potential for transdermal absorption and systemic availability of substantial amounts of salicylic acid in connection with the exposure resulting from the use of trolamine salicylate in sunscreens raises concerns about increased occurrence of the above-described serious adverse events (including salicylism and serious coagulation-related issues). The record also contains several significant data gaps that would need to be addressed to support a positive GRASE determination for trolamine salicylate. Start Printed Page 6221Given the safety concerns described above, however, conducting the clinical absorption testing recommended to address these gaps for use as a sunscreen raises ethical concerns.
C. Ingredients Proposed as Category III
The public record does not contain sufficient data to support a positive GRASE determination for cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, sulisobenzone, oxybenzone or avobenzone at this time. Accordingly, these ingredients are being proposed as Category III. In the sections that follow, we discuss our review of the available safety evidence for these ingredients and identify the existing data gaps.
1. Ingredients for Which the Record Contains Significant Data Gaps: Cinoxate, Dioxybenzone, Ensulizole, Homosalate, Meradimate, Octinoxate, Octisalate, Octocrylene, Padimate O, and Sulisobenzone
The most significant gaps in the administrative record exist for the following active ingredients: Cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, and sulisobenzone. We expect that data from all the types of studies described in section VII will need to be submitted to support general recognition of safety and effectiveness for each of these ingredients.
Only three of these active ingredients (homosalate (Ref. 81)), octinoxate (Refs. 81 to 84), and octisalate (Ref. 81), for example, appear to have been evaluated in human absorption studies, and most of the available absorption studies for these three ingredients had significant limitations. For example, the studies use a limited number of subjects or are based on only a single application of the sunscreen active ingredient to a limited area of the body. Even with this limited sunscreen exposure, some of these studies showed systemic availability of the active ingredient (octinoxate (Refs. 83 and 84)). None of these 10 ingredients has been studied in an adequate and well-controlled MUsT that would determine the amount of systemic exposure to the active ingredients under conditions of maximal use.
We note that a recent publication examining the relationship between melting point, molecular weight, and the transdermal delivery rates of the active ingredients in approved drug products shows that products containing active ingredients with melting points and molecular weights similar to many of these 10 sunscreen active ingredients are among those successfully delivered transdermally—and therefore available systemically (Ref. 85). This reinforces the potential for transdermal absorption of and systemic exposure to these sunscreen ingredients. The potential for such systemic exposure is a concern because the available data are inadequate to determine either the level of systemic exposure to these active ingredients or the potential unintended consequences of such exposure. Given the lack of chronic exposure toxicology data for these 10 ingredients—which makes an evaluation of the dermal and systemic effects of chronic use impossible—this is especially concerning. A number of these active ingredients have also shown hormonal effects in mammalian assays (homosalate (Refs. 86 to 92)) and padimate O (64 FR 27666 at 27671) and in in vitro and in vivo assays (homosalate (Refs. 86 to 92), octinoxate (Refs. 93 and 94),and octocrylene (Ref. 95). Although these findings are only preliminary, we do not have adequate DART studies to enable us to assess the impact of these potential hormonal effects on development and reproduction.
In addition, several of these 10 ingredients (homosalate (Refs. 81 and 84), octinoxate (Refs. 81 and 96 to 101), octisalate (Refs. 81, 84, and 101 to 105),octocrylene (Refs. 95 and 106), padimate O (Ref. 100), and sulisobenzone (Refs. 107 and 108)) have been studied in dermal penetration studies, which show (in general, with the exception of homosalate) that these ingredients permeate into the epidermis and/or dermis. The studies show that there are several factors (including vehicle composition and the presence of other active ingredients) that can influence, and potentially increase, the permeation and/or penetration of these ingredients.
Because the record does not currently contain sufficient data to support their safety, we are proposing that cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, and sulisobenzone are Category III ingredients. As previously noted, we expect that data from all the types of studies described in section VII will be needed to support general recognition of safety and effectiveness for these ingredients.
2. Ingredients for Which the Record Contains Fewer Data Gaps: Oxybenzone and Avobenzone
While the record does not currently contain sufficient data to support positive GRASE findings for oxybenzone and avobenzone, we have significantly more data for these two ingredients than for the ingredients discussed in the preceding section. To help facilitate submission of the remaining data, we describe the data gaps for these two ingredients in greater detail below.
a. Oxybenzone data. Although we located substantially more data on oxybenzone than on the ingredients discussed in section VIII.C.1, our review of the scientific literature, submissions to the sunscreen monograph docket, and postmarket safety data publicly available through FAERS revealed significant gaps in the data we expect to be necessary to support a positive GRASE finding for use of oxybenzone at a concentration of up to 6 percent in sunscreen products. The available literature includes studies indicating that oxybenzone is absorbed through the skin and can lead to significant systemic exposure, as well as data showing the presence of oxybenzone in human breast milk, amniotic fluid, urine, and blood plasma. The significant systemic availability of oxybenzone (and, as discussed further below, the lack of data evaluating the full extent of its absorption potential) is a concern, among other reasons, because of questions raised in the published literature regarding the potential for endocrine activity with systemic oxybenzone exposure. Accordingly, we expect that a positive GRASE finding for oxybenzone-containing sunscreens would require, among other things, both a MUsT showing the degree of oxybenzone absorption under maximal usage conditions and DART studies that fully investigate its potential endocrine-disrupting effects. We found neither in the existing record.
The record also lacks systemic and dermal carcinogenicity studies for oxybenzone; these (and toxicokinetic data) should also be provided to support a positive GRASE finding for this ingredient. Finally, the available literature also raises questions about the safety of use of oxybenzone-containing sunscreens in young children because of the potential for higher absorption and bioaccumulation of oxybenzone in this population. As discussed in further detail in the sections that follow, we invite input and comment on appropriate studies and/or age restrictions to address these pediatric issues.
b. Background of oxybenzone. Unlike zinc oxide and titanium dioxide, both of which are inorganic (or physical) UV filters consisting of metal oxides that primarily reflect or scatter UV radiation, Start Printed Page 6222oxybenzone is an organic (or chemical) filter, which absorbs UV radiation. It belongs to a class of aromatic ketones known as benzophenones and has a UV absorption profile covering both UVA and UVB wavelengths (Ref. 109). Because of its superior UVA coverage, oxybenzone was increasingly used through the early 1990s and ultimately replaced PABA in sunscreen products (Ref. 110). Use of oxybenzone in sunscreens increased when “PABA-free” sunscreens were introduced into the market because of recognition that PABA and its esters induced contact and photocontact allergic reactions (Ref. 110). As discussed below, however, evidence shows that oxybenzone also has contact allergenic and photoallergenic potential (Ref. 111). In addition to its use as a sunscreen active ingredient, oxybenzone is used in, among other things, perfumes, lipsticks, hair sprays, and conditioners as a photostabilizer and/or fragrance enhancer (Refs. 112 to 114).
c. Data showing transdermal absorption and significant systemic availability of oxybenzone. Data that have become available since publication of the Stayed 1999 Final Monograph suggest that the transdermal absorption of oxybenzone is high (Refs. 82, 115, and 116). One study involving sampling of plasma and urine following topical application of an oxybenzone-containing formulation showed absorption and significant systemic availability of oxybenzone (Ref. 82). In this study, 15 men and 17 women were dosed once daily, applying a 10 percent oxybenzone cream formulation to approximately 90 percent of the body's surface area for 4 days. The figures below illustrate the plasma and urine levels observed.
Start Printed Page 6223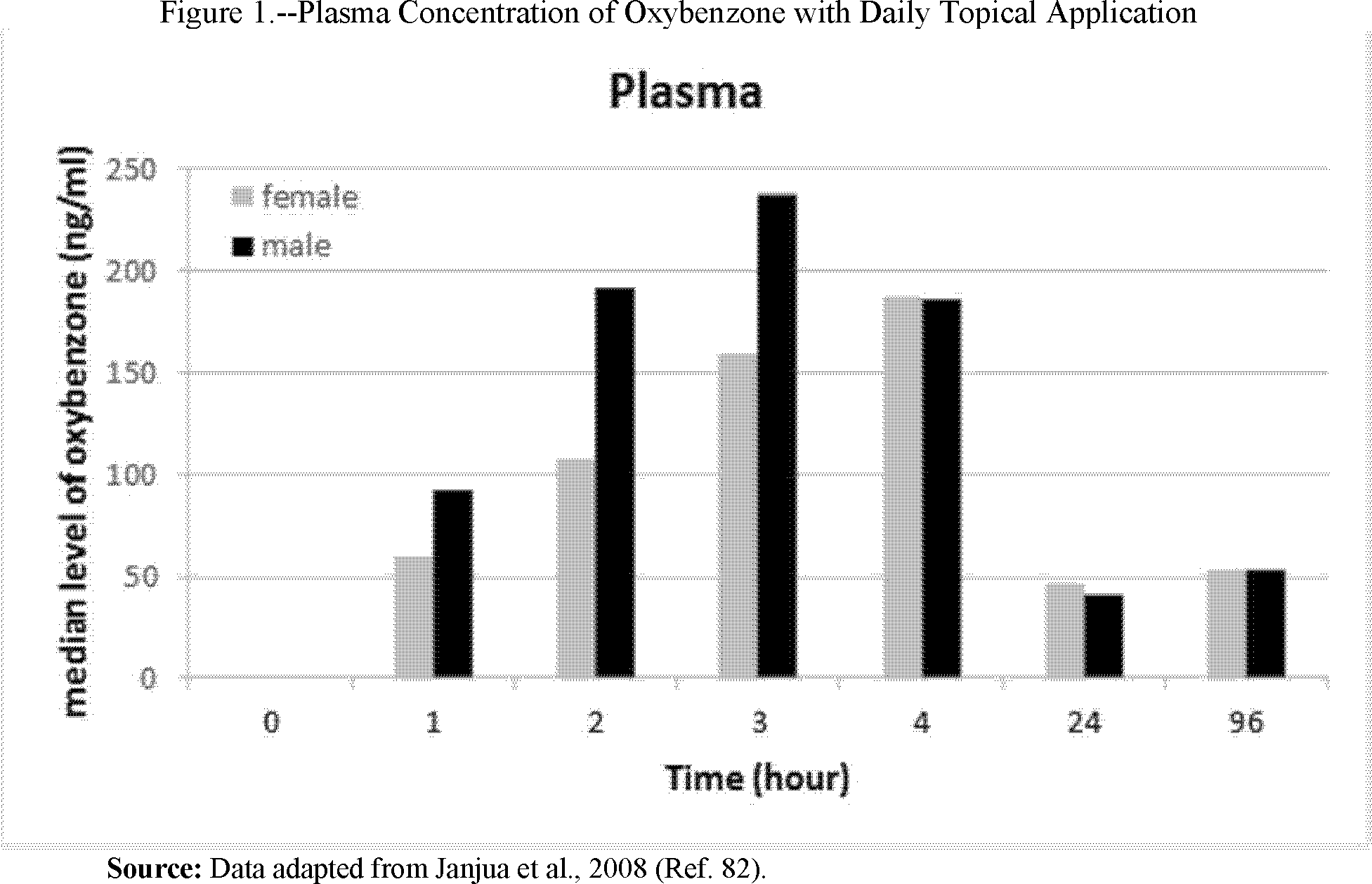

Although this study provides important information about the significant absorption potential of oxybenzone, it does not obviate the need for a MUsT. Among other things, once-daily application may result in substantially lower systemic exposure than application at least every 2 hours (as sunscreen labeling directs). This difference in application frequency is a particular concern given that the data show oxybenzone levels would still be increasing at the time of reapplication if a 2-hour application window were observed. Additionally, the cream formulation used in the study was not formulated as a sunscreen product and may have contained ingredients not typically used in sunscreen formulations, and/or lacked other ingredients typically present. Because the formulation can have an impact on absorption, the absorption results produced by the study may not reflect absorption levels that would result from actual use of oxybenzone-containing sunscreen products.
Another study, which evaluated the transdermal absorption of a marketed sunscreen containing 4 percent oxybenzone in 16 women and 9 men, showed prolonged systemic availability of oxybenzone following topical exposure (Ref. 116). In this study, which was designed to evaluate the effects of UV radiation on oxybenzone absorption, the sunscreen was applied to study subjects twice daily for 5 days. Although the study concluded that UV exposure did not significantly affect the urinary excretion of oxybenzone, it provided further evidence of the systemic availability of oxybenzone following topical application and showed that renal excretion of oxybenzone continued for 5 days after the last application of the sunscreen. Although the use of a commercial sunscreen formulation, and twice- rather than once-daily sunscreen application are improvements over the formulation and application frequency used in the previous study, twice-daily application remains insufficient to approximate the recommended application frequency of sunscreen products in real-world use. Furthermore, because the study used a sunscreen with 4 percent rather than the full 6 percent concentration of oxybenzone eligible for the sunscreen monograph, its results may not fully reflect the absorption that would result from use of a 6 percent oxybenzone-containing product. To properly characterize the potential for absorption of oxybenzone in sunscreen products and to determine a margin of safety for use of oxybenzone at up to 6 percent in sunscreen products, we expect that a MUsT will be needed.
d. Inadequate data on oxybenzone's developmental and reproductive toxicity. The significant systemic availability of oxybenzone following topical application and the lack of data fully characterizing its absorption levels are concerns, among other reasons, because of literature suggesting that oxybenzone may have endocrine activity (see, e.g., Refs. 88, 92, and 117). Dermal exposure to oxybenzone (in acetone) in rats and mice and oral feeding of oxybenzone to rats and mice resulted in reduced sperm density in males in 13-week general toxicity studies conducted by the National Toxicology Program (NTP) (Ref. 118). In female rats and mice, increased estrous cycle length was observed in 13-week oral feeding studies.[31] Importantly, the actual effects of oxybenzone on female fertility were not evaluated. In a preliminary dose range-finding pre- and postnatal development study in rats, findings in male offspring indicated that cells in the testes undergoing programmed cell death were increased in all oxybenzone-exposed animals and that numbers of spermatocytes in the testes were markedly reduced after oral feeding at oxybenzone (Ref. 119). Although these findings are notable, they are all derived from dermal studies Start Printed Page 6224with oxybenzone in acetone and oral feeding studies of oxybenzone; these methods of exposure could potentially lead to higher levels of systemic exposure to oxybenzone than with sunscreen use. Accordingly, a MUsT and toxicokinetic data are needed to determine the relevance of these findings to human use of oxybenzone as a sunscreen active ingredient.
In humans, the endocrine effects of oxybenzone have been studied with inconclusive results (see, e.g., Refs. 83, 120, and 121). In biomonitoring studies of pregnant and lactating women, oxybenzone has been detected in breast milk, amniotic fluid, and urine samples (Ref. 83, 120, and 121). High levels of oxybenzone in the urine of mothers have been associated with: (1) Decreased birth weight in girls and (2) increased birth weight and head circumference in boys, both of which can be indications of endocrine effects (Ref. 83). This association is particularly concerning given the widespread exposure of the U.S. population to oxybenzone. Estimates suggest that oxybenzone (from all sources) is present in the urine of 97 percent of the U.S. population, and that oxybenzone concentrations are higher in women than in men (possibly because women are more likely to use sunscreen and other personal care products containing oxybenzone, leading to greater cumulative exposure) (Ref. 83 and 115).
Because current data suggest that oxybenzone may affect the human endocrine system, FDA believes that a positive GRASE determination for oxybenzone would require that its potential toxicities have been fully explored, including through DART studies (fertility and early embryonic studies in rodents, embryofetal development studies in rodent and nonrodent species, and pre- and postnatal development studies in rodents). In addition, as noted below, toxicokinetic data are needed to interpret these studies. We note that, if the results of DART studies do not resolve the concerns raised in the literature relating to potential endocrine disruption, it may still be possible to resolve these concerns through additional testing.[32] In addition, because of the potential risk posed by metabolites of oxybenzone (existing reports suggest that some oxybenzone metabolites are more hormonally active than the parent drug (Ref. 109)), we recommend that the analytical method used in the MUsT be validated for both the parent and the metabolites of interest (Ref. 115) to support a positive GRASE finding for this ingredient. The results from the metabolite study will inform whether additional nonclinical studies assessing oxybenzone's metabolites should be conducted to support its safety. We note that the NTP is currently conducting additional DART studies on oxybenzone (although their embryofetal studies do not appear to include an assessment in a nonrodent species) (Ref. 122).[33]
e. Inadequate carcinogenicity and toxicokinetic data for oxybenzone. High population exposure to oxybenzone, coupled with a lack of carcinogenicity testing for this ingredient, caused the National Cancer Institute to nominate oxybenzone for toxicology testing in 1979 (Ref. 123). The NTP reports that 2-year oral (dosed feed) carcinogenicity studies in rats and mice are in a draft report stage, but results are not yet publicly available (Ref. 122). In addition, no reports of either ongoing or planned dermal carcinogenicity studies for oxybenzone have been published. To support a positive GRASE finding for oxybenzone, carcinogenicity data from well-conducted dermal and systemic carcinogenicity studies should be provided. Toxicokinetic data in rodents (oral and dermal) and rabbits (oral) are also recommended; these data could be obtained from either stand-alone studies or as part of DART and dermal carcinogenicity studies.
Our search of the available literature also revealed information suggesting that oxybenzone may generate reactive oxygen species (ROS) [34] in the presence of UV light, but that this issue, and the harms associated with it, have not been fully explored (Ref. 124). We invite comment and input on the extent to which ROS generation is a concern for sunscreens containing oxybenzone and whether additional data on this topic are needed.
f. Dermal safety of oxybenzone. The available data indicate that oxybenzone (at concentrations up to 6 percent) has a favorable safety profile with respect to irritation and sensitization potential. For example, the North American Contact Dermatitis Group conducted an analysis of patients who were patch tested for allergies between 2001 and 2010 (see, e.g., Ref. 125). From 2001 to 2008, oxybenzone was tested at 3 percent; from 2009 to 2010, the concentration used for the test was increased to 10 percent. Of the 23,908 patients patch tested, only 82 patients (0.34 percent) had positive test patch results with oxybenzone. In addition, a search of FAERS for case reports of hypersensitivity reactions to oxybenzone-containing sunscreen products resulted in only 31 cases (4 with anaphylaxis) between 1988 and 2011. Because sufficient data exist to make a determination, we do not consider additional dermal irritation or sensitization studies to be necessary to support a positive GRASE finding for oxybenzone up to 6 percent. As is customary in clinical trials, however, we recommend that dermal safety data for oxybenzone be collected during MUsT studies.
Nevertheless, the overwhelming majority of results from available studies (see, e.g., Refs. 125 to 136) addressing allergic contact dermatitis for oxybenzone show that oxybenzone is an allergen for persons with preexisting skin conditions. Because the evidence establishing oxybenzone as a photoallergen in individuals with photosensitivity is clear, no further dermal photosafety studies to characterize this risk are needed. However, if we were to receive adequate data to support a positive GRASE finding for oxybenzone, we would consider requiring labeling language to address the risk of allergic reactions associated with oxybenzone use. We invite comment on whether such labeling should be required for sunscreens containing oxybenzone and, if so, what that labeling should entail.
g. Safety questions regarding use of oxybenzone in pediatric populations. Sunscreens are currently labeled for use in children as young as 6 months old. The available literature, however, includes several publications that raise concerns about the use of sunscreens containing oxybenzone in young children. Among these publications is a 2006 report from the Swedish Research Council noting that children under the age of 2 years old have not fully developed the enzymes believed to metabolize oxybenzone (Ref. 137), which suggests, in theory, that small children may not be able to eliminate oxybenzone as easily as adults. The possibility for bioaccumulation in children, taken together with the potential increased absorption of oxybenzone in young children (due to their greater body surface-area-to-weight ratio) and the potential harms associated with absorption discussed above, Start Printed Page 6225militates in favor of caution when using oxybenzone products in young children. Accordingly, we are seeking any existing pediatric data on the safety of oxybenzone use in children under 2 years old. We are also requesting input on: (1) Whether additional data on the safety of oxybenzone use in young children is necessary to support the use of oxybenzone-containing sunscreens in children under 2 years of age (taking into consideration the practical hurdles involved in conducting studies in children of this age) or (2) whether sunscreen products containing oxybenzone should instead be contraindicated for use in children younger than 2 years (given, among other things, the availability for use as sunscreen active ingredients of physical UV filters like titanium dioxide and zinc oxide, which do not raise the same questions about safe use in young children).
h. Conclusion. Given the available data showing significant transdermal absorption and systemic availability of oxybenzone, as well as the potential for endocrine activity, we propose that oxybenzone is not GRASE for use in sunscreens without further data. As described above, a MUsT should be conducted to fully characterize the absorption of oxybenzone and to calculate a margin of safety for human use. As part of the MUsT, we believe that a study of oxybenzone's metabolites in humans is also necessary; the results of this study will inform whether additional nonclinical studies with metabolites are needed to address potential endocrine effects. Given that oxybenzone demonstrates significant systemic absorption, FDA believes that data on carcinogenicity (both systemic and dermal) and developmental/reproductive toxicity are likely to be needed to support the safety of this ingredient, as are toxicokinetic data to bridge between animal and human data. We seek any existing data on the pediatric safety of oxybenzone. We also seek comment on whether additional safety data are needed to support the use of sunscreens containing oxybenzone on children under 2 years of age, as well as comment on whether these sunscreens should be contraindicated for use in this population. We note that, because of the risk of allergic reactions associated with oxybenzone use, if we receive adequate data to support a positive GRASE finding for oxybenzone, we may require labeling to address this risk. We seek comment on whether such labeling should be required for sunscreens containing oxybenzone and, if so, what such labeling language should entail.
In summary, table 3 shows the additional studies that FDA anticipates would be necessary to support a positive GRASE finding for sunscreens containing oxybenzone.
Table 3—Summary of Recommendations: Studies for Oxybenzone Up to 6 Percent
Safety studies FDA proposes are necessary to support a GRASE determination Additional studies or data necessary? Pharmacological Studies: Human absorption (MUsT) (including metabolite study in humans) Yes. Nonclinical Safety Studies: Toxicokinetics Yes. Dermal Carcinogenicity Yes. Systemic Carcinogenicity Yes. DART: 1 Yes. Fertility and early embryonic development. Embryofetal development in two species (rodent and non-rodent). Prenatal and postnatal development. Clinical Safety Testing: Skin irritation and sensitization No. Skin photoallergenicity and phototoxicity No. Pediatric studies Seeking input on whether additional studies or contraindication are necessary to support the safety of sunscreens containing oxybenzone for children under 2 years of age. 1 As noted above, if DART studies do not resolve the concerns raised in the literature relating to potential endocrine disruption, it may be possible to resolve these concerns through additional testing. i. Avobenzone data. Our review of the available scientific literature, submissions to the sunscreen monograph docket, and publicly available FAERS data also revealed significant gaps in the data we expect to be necessary to support a finding that avobenzone (at up to either 3 percent or 5 percent, as discussed below) is GRASE for use in sunscreens. Most critically, we encountered no studies examining the absorption of avobenzone in vivo, and those in vitro studies we located had several weaknesses that limit their usefulness in assessing the potential absorption of avobenzone from formulated sunscreen products. This is a concern given that, as explained in further detail below, certain of avobenzone's chemical properties suggest that sunscreen products containing avobenzone have a potential for absorption. There are also other gaps in the record, including (as discussed below) dermal carcinogenicity data, toxicokinetic data, and—potentially, depending on the outcome of MUsT studies assessing the absorption of avobenzone—systemic carcinogenicity and additional DART studies. Accordingly, we propose to find that avobenzone is Category III.
j. Background of avobenzone. Avobenzone, like oxybenzone, is an organic (chemical) UV filter. Because avobenzone primarily absorbs radiation in the UVA portion of the UV spectrum, it is typically combined with another sunscreen active ingredient that provides protection in the UVB range. Avobenzone exhibits greater photoinstability than other UV absorbers; the available evidence shows that avobenzone degrades quickly upon exposure to sunlight, which can cause its efficacy to be decreased by between 50 and 90 percent after 60 minutes of exposure to sunlight (Refs. 138 and 139).[35] To address this, avobenzone is typically combined with a photostabilizer to prevent rapid photodegradation (Refs. 138 and 139).
Start Printed Page 6226k. Potential for absorption of avobenzone. Although avobenzone is not soluble to any great extent in water, it is soluble in organic solvents. These include oils (which are present on human skin), alcohols, and other substances regularly included in sunscreen product formulations. Although this solubility is not enough, by itself, to determine whether transdermal absorption will take place, it is a necessary precondition (Ref. 140). In addition, like the 10 active ingredients described in section VIII.C.1, avobenzone's melting point and molecular weight are similar to those of active ingredients in approved drug products that are successfully delivered transdermally and therefore available systemically (Ref. 85). As with the 10 sunscreen active ingredients previously discussed, this suggests a potential for transdermal absorption of avobenzone.
l. Lack of adequate data on transdermal absorption of avobenzone. Nevertheless, our review of the available literature on avobenzone failed to produce any studies evaluating the in vivo absorption of avobenzone at 3 percent or higher under (or even approaching) maximal usage conditions. While we were able to locate a few studies evaluating avobenzone's absorption in vitro, these studies had a number of weaknesses that significantly limited the conclusions that could be drawn from them.
The first in vitro study we located evaluated the penetration—through excised human skin—of five sunscreen ingredients (including avobenzone) that had been diluted in mineral oil and water (Ref. 100). The study used a static cell technique. As discussed in section VIII.D, in a static cell study, the test product (here, a sunscreen/mineral oil/water formulation) is placed on the upper side of a membrane (here, the excised skin) in the open donor chamber of a static cell, and a sampling fluid is placed on the other side of the membrane in a receptor cell. Diffusion of the ingredient (here the avobenzone) from the topically applied product to and across the membrane is monitored by examining sequentially collected samples of the receptor fluid. To ensure that all transdermal penetration of the ingredient that takes place is fully reflected in the receptor fluid, the receptor fluid must be optimized for absorption (in other words, sink conditions must be created in the fluid).
In this study, the use of skin as the membrane in the system allowed for an evaluation of the presence and depth of permeation via skin stripping—the sequential application and removal of adhesive tape to the skin samples. However, it is unclear whether the receptor phase of the study created adequate sink conditions. In addition, the formulations used in the study (which, as noted previously, consisted of only water, mineral oil, and the sunscreen ingredient) did not contain any of the other types of excipients (such as emollients, stabilizers, or solubilizers) that can also function as permeation/absorption enhancers and that are typically present in sunscreen product formulations. The study results showed that there was avobenzone present in the stratum corneum, the epidermis, and the viable dermis of the skin used as the membrane, but not in the receptor fluid. Although the lack of avobenzone in the receptor fluid is encouraging, the other characteristics of the study limit its value in assessing the actual absorption potential of avobenzone used in sunscreen products.
The second in vitro study (Ref. 141) we located suffered from similar limitations. This study assessed the avobenzone permeation observed using a static cell (as generally described above), and then took the skin from the static cell and subjected it to multiple rounds of tape stripping to assess the presence of avobenzone at various levels of the skin. Following tape stripping, the skin was subjected to digestion (i.e., the skin sample was subjected to a chemical treatment that breaks down the cell membranes to release any sunscreen that might be either bound to proteins or bound up in the cells).
The study results showed significant retention of avobenzone in the stratum corneum, a lesser amount in the epidermis, and none in the dermis or receptor fluid. Like the previous study, however, the test material used in this study did not include any of the permeation enhancers typically included in commercial sunscreen formulations. It is also unclear whether sink conditions existed in the receptor phase of the study.
The final in vitro study used a static cell to evaluate the transdermal penetration of six sunscreen formulations collected from a health spa that marketed its own line of skin care products (Ref. 96). This study improved on the design of the previous two studies in several respects. First, the receptor fluid contained ethanol, a permeation enhancer often used in sunscreen products, which produced sink conditions in the receptor phase. Secondly, to create favorable conditions for absorption, the products were applied at a thickness of 20 mg/square centimeters (cm2) on the skin's surface (i.e., 10 times the skin loading typically expected (Refs. 142 and 143)). In addition, the study's use of commercially marketed sunscreen formulations (which, as discussed above, typically contain multiple permeation/absorption-enhancing excipients) more accurately reflects the absorption potential of marketed sunscreen products.
Despite these improvements, the usefulness of the study was limited by its use of an analytical method that prevented the detection of any avobenzone absorption below 100 ng/mL. This level of absorption is hundreds of times higher than what is relevant for our considerations in assessing the acceptable absorption level from a topically applied product. The concentration of avobenzone used in the study (ranging from 0.2 percent to 1 percent) is also significantly lower than what is relevant for our current consideration of maximum concentration of this ingredient. Although avobenzone was only absorbed to a very small extent (between 3 percent and 3.96 percent) under these study conditions, these weaknesses in the study's design significantly limit the conclusions that can be reached from its results.
Given that avobenzone's chemical properties suggest that it has a potential for transdermal absorption in sunscreen products, the lack of adequate data assessing its absorption in realistic sunscreen formulations is a concern. We therefore expect that a MUsT demonstrating the degree of absorption of avobenzone into the human body under maximal use conditions will be needed to support a positive GRASE determination for sunscreens containing avobenzone. Further, in light of the above-described data showing avobenzone's photoinstability, we also expect that, if sufficient data are provided to support the safety of avobenzone, any future sunscreen monograph including avobenzone as an active ingredient will include the limitation that avobenzone is not GRASE for use in sunscreen products unless it has been photostabilized (via use of a photostabilizing UV filter or other photostabilizing ingredient/mechanism) to prevent its photodegradation and (among other concerns) the attendant reduction in avobenzone efficacy.
Because photodegradation can reduce the amount of avobenzone absorbed transdermally, we also expect that a MUsT sufficient to support the general recognition of safety of avobenzone for sunscreen use would need to test formulations of avobenzone that include a photostabilizer. Including Start Printed Page 6227photostabilizers in MUsT formulations will allow for accurate assessment of absorption levels in final formulated sunscreen products containing avobenzone. This proposal is consistent with our general recommendation that materials evaluated under the MUsT paradigm represent real-world sunscreen formulations, rather than overly simplified solutions that fail to replicate the absorption potential of marketed formulations. As noted in section VII.B.4, we encourage sunscreen manufacturers to discuss their MUsT protocol with FDA before beginning the trial.
m. Data supporting dermal safety of avobenzone. The available clinical dermal studies indicate that avobenzone at concentrations up to 5 percent have a favorable safety profile with respect to potential irritation, sensitization, and photosafety. In 2009, in conjunction with a citizen petition [36] (L'Oreal Petition, Docket No. FDA-1978-N-0018-0675) asking FDA to take action to permit the marketing of sunscreen products containing avobenzone up to 5 percent, L'Oreal USA Products, Inc. (L'Oreal) submitted nine human repeat insult patch, phototoxicity, and photoallergy studies with six different sunscreen formulations containing avobenzone (3.4 percent, 4 percent, or 5 percent). The studies showed that the formulations were well tolerated for topical use (i.e., essentially non-allergenic, non-irritating, and non-sensitizing, with mild to moderate reactions occurring only rarely) (L'Oreal Petition).[37] A separate search of the available scientific literature on the clinical safety of avobenzone did not reveal anything to undermine these findings. Although the available literature included a small number of reports of contact irritation and photosensitization in connection with avobenzone-containing products, details about the composition of the formulations at issue (and the concentrations of avobenzone) were frequently missing from the literature, making it difficult to determine the cause of these responses. A small number of serious hypersensitivity reports for sunscreens containing avobenzone were also located in FAERS. Because the sunscreens at issue usually contained three or more active ingredients, however, it is difficult to determine what caused the reaction. Because sufficient data exist to make a determination, we do not consider additional dermal clinical studies (including photosafety, irritation, or sensitization studies) to be necessary to support the safety of this ingredient for sunscreen use at up to 5 percent. As is customary in clinical trials, however, we recommend that dermal safety data for avobenzone be collected during MUsT studies.
n. Other nonclinical safety studies for avobenzone. Dermal carcinogenicity studies have not been conducted for avobenzone. The available data on the permeation of avobenzone suggest that it may permeate into at least the dermis and epidermis, which means that it is possible for avobenzone to impact skin tumor development. We therefore expect that dermal carcinogenicity studies will be necessary to support a positive GRASE finding for sunscreens containing this ingredient. Available embryofetal development studies in rats and rabbits did not reveal any findings of concern. However, our review of the nonclinical data for avobenzone [38] also revealed that toxicokinetic data following repeat-dose exposure will be needed to interpret pivotal nonclinical safety studies (including the embryofetal development studies in rats and rabbits) once the MUsT data become available. (As explained in section VII.B.4, these data are used to compare drug levels achieved in animal studies with those observed in humans under maximal exposure conditions.) In addition, if results of a MUsT demonstrate that there is significant systemic absorption of avobenzone, additional fertility and early embryonic development and prenatal and postnatal development studies in rats will be needed to support a positive GRASE finding. Depending on the results of the MUsT, systemic carcinogenicity studies may also be needed.
o. Avobenzone in combination with other sunscreen active ingredients. As noted in section III.B, our finding in the Stayed 1999 Final Monograph that avobenzone was GRASE for use in sunscreens would have allowed its combination only with certain other sunscreen active ingredients (64 FR 27666 at 27688) because we did not have targeted evidence to support the safety and effectiveness of avobenzone when combined with the remaining active ingredients. We believe this limitation was inconsistent with the approach to evaluating sunscreen combinations that the Agency has generally taken throughout the OTC Drug Review for sunscreens. For this reason, unless evidence is submitted to suggest that there is a safety or efficacy concern associated with the combination of avobenzone with another active ingredient, we expect to conclude that a positive GRASE determination for avobenzone will support its use in sunscreens either alone or in combination with all other sunscreen active ingredients.
p. L'Oreal request to increase concentration of avobenzone to 5 percent. Avobenzone is currently listed in the Stayed 1999 Final Monograph at concentrations up to 3 percent. As described earlier, in 2009 FDA received a citizen petition from L'Oreal requesting, among other things, that we amend the sunscreen monograph to increase the allowable level of avobenzone to 5 percent (L'Oreal Petition at 1). In the Stayed 1999 Final Monograph, the Agency determined that avobenzone at concentrations up to 3 percent is an effective sunscreen active ingredient. We now likewise conclude that the record contains sufficient information to satisfy the effectiveness prong of the GRASE standard for sunscreens containing avobenzone at concentrations up to 5 percent.
As described above, data submitted with that L'Oreal Petition were sufficient to establish that avobenzone at a concentration of up to 5 percent has a favorable safety profile with respect to potential irritation, sensitization, and photosafety. To support a finding that avobenzone at concentrations up to 5 percent is GRASE for use in sunscreens, however, FDA expects that a MUsT evaluating the transdermal absorption of avobenzone up to 5 percent, as well as dermal carcinogenicity studies and toxicokinetic data for avobenzone at a concentration of at least 5 percent, will also be needed. Depending on the outcome of the MUsT, we may also need systemic carcinogenicity data and additional DART studies, including fertility and early embryonic development, and pre- and postnatal development studies in rats for avobenzone at 5 percent. The record does not currently include any of these data. However, if FDA were to receive sufficient data to support a positive GRASE finding for avobenzone up to 5 percent, we would expect to include Start Printed Page 6228avobenzone at this percentage in a final sunscreen monograph.
q. Conclusion. Given that: (1) Avobenzone's organic solubility, molecular weight, and melting point suggest it has a potential for transdermal absorption; (2) there is a lack of available data on the transdermal absorption of avobenzone in vivo (including under maximal use conditions); and (3) there are limitations in the available in vitro studies assessing avobenzone absorption, we expect that a properly designed MUsT will be necessary to support a positive GRASE finding for avobenzone use in sunscreens. We expect that, to be GRASE for sunscreen use, avobenzone will need to be photostabilized to address its potential for degradation, and we therefore expect that any future sunscreen monograph including avobenzone as an active ingredient will include the limitation that avobenzone is not GRASE for use in sunscreen products unless it has been photostabilized to prevent its photodegradation. In addition, we believe that an adequate MUsT evaluating the absorption potential of avobenzone will need to include a photostabilizer to ensure that the potential transdermal absorption of avobenzone from avobenzone-containing sunscreens is accurately assessed.
We also expect that dermal carcinogenicity and toxicokinetic data will be necessary to support a positive GRASE finding for sunscreens containing avobenzone. Depending on the outcome of a MUsT assessing the absorption of avobenzone, systemic carcinogenicity testing and additional DART studies, including fertility and early embryonic development and pre- and postnatal development studies in rats may be needed as well. We will also determine the extent to which additional DART studies may be needed based on the results of the MUsT. Depending on the results of the nonclinical and pharmacology studies for this ingredient and the safety margin that is calculated from these results, pediatric studies for avobenzone may also be needed to support the use of sunscreens containing avobenzone in pediatric populations.
Table 4—Summary of Recommendations: Studies for Avobenzone Up to 3 (or 5) Percent
Safety studies FDA proposes are necessary to support a GRASE determination Additional studies or data necessary? Pharmacological Studies: Human absorption (MUsT) (including metabolite study in humans) Yes. Nonclinical Safety Studies: Toxicokinetics Yes. Dermal Carcinogenicity Yes. Systemic Carcinogenicity Dependent on results of the MUsT. DART: Fertility and early embryonic development Dependent on results of the MUsT. Embryofetal development in two species (rodent and non-rodent) No. Prenatal and postnatal development Dependent on results of the MUsT. Clinical Safety Testing: Skin irritation and sensitization No. Skin photoallergenicity and phototoxicity No. Pediatric studies Pediatric studies may be required depending on the outcome of the MUsT. D. Anticipated Final Formulation In Vitro Permeation Testing
As noted earlier, a final sunscreen monograph will need to set out the conditions under which any product marketed pursuant to it would be GRASE and not misbranded. Variations among individual sunscreen product formulations—in particular, characteristics of the specific vehicle (e.g., the cream, lotion, or oil) in which active ingredients are delivered—can affect the transdermal absorption of sunscreens, and thus, have an impact on their safety and effectiveness. To address this, FDA currently requires final formulation testing of OTC sunscreen products to support labeled claims regarding their effectiveness—namely, testing for SPF value as well as broad spectrum protection and water resistance where those attributes are claimed in product labels.[39] For purposes of this proposed rule, we use the term final formulation testing to refer to testing conducted on the sunscreen product formulation to be marketed. Our expectation is that final formulation testing would also generally be necessary to ensure that the active ingredient in any given sunscreen formulation permitted under the monograph would not be systemically absorbed beyond the amount FDA determined to be safe.
The discussion that follows provides FDA's thinking about such testing of final formulations, which we anticipate requiring in the future for sunscreen products marketed under the sunscreen monograph (unless FDA determines that the ingredient or ingredients contained in the product are unlikely to be absorbed through the skin). Because this testing would not be required for sunscreens containing only those active ingredients proposed here as Category I (zinc oxide and titanium dioxide), FDA has not yet reached a final determination as to the particular parameters that might be required for such final formulation testing. We anticipate that we may specify final formulation testing requirements in the monograph in the future, however, as active ingredients that we are now proposing as Category III may be included in the monograph in the future if FDA receives data supporting their GRASE status. Final formulation testing requirements applicable to such ingredients would be established on an ingredient-specific basis, taking into consideration the data provided to Start Printed Page 6229support a positive GRASE determination for the specific ingredient (for example, whether any safety signals are detected in well-conducted nonclinical carcinogenicity and DART studies). We encourage interested parties to provide information and comment for each sunscreen active ingredient that is relevant to establishing this kind of final formulation testing for each active ingredient.
FDA's expectation is that this testing would not generally call for an in vivo study. Instead, FDA expects that the conditions of marketing specified for sunscreens containing a given active ingredient would require manufacturers to perform in vitro permeation testing before marketing each sunscreen formulation containing that ingredient. Consistent with the approach for SPF and broad spectrum final formulation testing set forth in § 201.327 (for proposed changes to § 201.327, see codified section of this document), FDA anticipates that it would not review the results of the in vitro permeation testing before product marketing. Rather, FDA expects that any future conditions pertaining to final formulation in vitro permeation testing in the sunscreen monograph would include a requirement that manufacturers maintain records of this testing, and that those records be available for FDA inspection upon request.
FDA anticipates establishing a standard control formulation for each sunscreen active ingredient to be used in the in vitro permeation testing of products containing that ingredient. The standard control formulation would be the formulation that produces the highest in vivo absorption in the MUsT. The results of in vitro permeation testing using this control formulation would then be used as a bridge to a corresponding level of in vivo absorption from the MUsT that is used to establish the safety margin for the ingredient. Then, FDA anticipates establishing conditions to ensure that final formulation in vitro permeation testing would be conducted for each formulation intended to be marketed, using the specified vertical diffusion cell described below. The results of the in vitro permeation testing of each final formulation would be compared to the absorption found in the standard control formulation for the active ingredient it contains.
In vitro permeation testing is a methodology that has been used in dermal formulation development for over 30 years and, as used here, specifically refers to use of the “Vertical Diffusion Cell” (Ref. 144). A vertical diffusion cell is comprised of three major units: (1) An upper chamber (into which the sunscreen formulation is placed); (2) the rate-limiting membrane (the prepared human skin); (3) and the lower chamber/fluid channel (containing a receptor fluid that is evaluated to determine how much of the sunscreen it “receives”) (Refs. 145 to 147). The vertical diffusion cell system has been commercialized and is available as both single and multiple unit models that can be automated.
Other relevant parameters FDA expects to consider in establishing future requirements for in vitro permeation testing include (among other things) the thickness and integrity of collected skin, storage conditions used for collected skin, receptor fluid composition, skin and receptor fluid temperature, the number of skin samples (and donors) used, study duration, sampling period, application method, and number of experimenters.
We note that if a final sunscreen formulation contains a combination of sunscreen active ingredients, FDA anticipates requiring that this final formulation be tested against the standard control formulations for each of the sunscreen active ingredients it contains. As noted above, a standard control formulation might not be specified for (and final formulation in vitro permeation testing might not be necessary to establish safety for) a sunscreen containing a particular active ingredient if FDA determines that the ingredient is unlikely to be absorbed through the skin. As mentioned above, we therefore do not propose to require final formulation in vitro permeation testing for sunscreen formulations containing only zinc oxide and/or titanium dioxide.
In cases in which such testing is required, FDA anticipates that if the in vitro permeation of each sunscreen active ingredient in the final formulated product is equal to or less than the value obtained from in vitro permeation testing of the standard control formulation for that active ingredient, the product's safety margin would be considered to fall within the parameters judged to be GRASE, and thus to support marketing of the formulation. However, if the in vitro permeation of the active ingredient from the specific final formulation is greater than the value obtained from in vitro permeation testing of the standard control formulation for that active ingredient, FDA anticipates that the drug product(s) using that formulation would not be considered GRASE. In that situation, the sponsor would have the option to either: (1) Reformulate the product and conduct in vitro permeation testing to establish that the reformulated product satisfies the final formulation in vitro permeation testing requirements set out in the sunscreen monograph or (2) seek NDA approval for the new formulation.
IX. Additional Proposed Conditions of Use
A. Proposed Requirements Related to Dosage Form
OTC sunscreens have been marketed in a variety of dosage forms over the years. Responding in part to the growing market acceptance of spray sunscreens, on June 17, 2011, FDA issued an ANPR addressing sunscreen dosage forms (Dosage Forms ANPR) (76 FR 35669, June 17, 2011). The ANPR identified dosage forms considered eligible or ineligible for review and potential inclusion in the OTC sunscreen monograph, based on FDA's knowledge, at that time, of their history of marketing before the OTC Drug Review began in 1972. It also solicited specific information about the safety, effectiveness, and directions for use of spray sunscreens.
1. Summary of Eligible and Ineligible Dosage Forms
In this proposed rule, FDA is confirming that the following dosage forms identified in the Dosage Forms ANPR are eligible for review and potential inclusion in the OTC sunscreen monograph based on their history of sunscreen marketing before 1972: Oil, lotion, cream, gel, butter, paste, ointment, stick, spray, and powder. With the exception of powder, FDA proposes that sunscreens in these dosage forms are GRASE subject to certain conditions described below and elsewhere in this proposed rule. We note that sunscreen powders were classified as ineligible for review in the Dosage Forms ANPR because, at that time, we were unable to identify any sunscreen products in powder form that were marketed before the OTC Drug Review began. Based on marketing data submitted to the ANPR docket and in a related citizen petition (Docket No. 1978-N-0018-0741), we have determined that the powder dosage form is eligible to be considered for inclusion in the OTC sunscreen monograph. However, as described in section IX.A.4, we tentatively conclude that additional safety and efficacy data will be necessary to classify sunscreens in the powder dosage form as GRASE and include them in the final monograph. We are proposing that sunscreens in all dosage forms other than those identified as eligible for consideration above—Start Printed Page 6230including wipes, towelettes, body washes, and shampoos—are new drugs because we did not receive data showing that they were marketed prior to 1972.
2. Overview of Comments on the Dosage Forms ANPR
FDA received a total of 14 comments on the Dosage Forms ANPR. Six of the comments provided no new data, but generally supported the advantages of spray sunscreens, agreed with the need to address concerns about spray sunscreens' performance and/or safety (especially when used on children), opined that existing SPF methods would not need to be modified for sprays, or (in most cases) agreed with FDA's suggested directions for use. Other comments argued for the inclusion of additional dosage forms identified as ineligible in the Dosage Forms ANPR, but failed to provide supporting marketing data. One comment contained marketing information showing that sunscreen products in powder form, which we had previously identified as ineligible for the monograph, had been marketed in the United States before 1972. The remaining comments (all from industry) provided data and information that directly or indirectly addressed questions raised in the Dosage Forms ANPR concerning the safety, effectiveness, and labeling of spray sunscreens. These comments are discussed in sections IX.A.3 and IX.A.4 below.
3. Safety and Effectiveness of Spray Sunscreens
As we recognized in the Dosage Forms ANPR, compared to traditional lotions, oils, and the like, spray sunscreens raise potential concerns of both safety and efficacy that FDA must consider in determining whether sunscreens in the spray dosage form would be GRASE. With respect to efficacy, FDA must consider factors such as whether spraying sunscreen rather than applying it by hand provides effective coverage on exposed skin, how consumers use spray products, and whether current test methods for SPF and broad spectrum protection can be relied on for adequate labeling of spray products. With respect to safety, spray sunscreens raise the question of potential harm from inhalation of sunscreen components as well as potential flammability risks.
a. Characteristics of sunscreen spray products. Spray sunscreens use varying technologies to package and deliver a sunscreen formulation as an aerosol spray, i.e., an airborne suspension of fine droplets or particles. In some spray products, the sunscreen formulation is mixed in a canister with a liquefied gas propellant that supplies the force to generate an aerosol containing both dissolved sunscreen formulation and propellant upon activation of a valve system. There are also pump spray sunscreen products that are not packaged under pressure but generate spray by applied mechanical force without the need for a propellant. Many currently marketed spray sunscreen products use a delivery technology referred to as a bag-on-valve system, in which the sunscreen formulation is contained in a bag with an attached valve inside a canister filled with propellant, so as not to mix the sunscreen formulation and propellant ingredients. For purposes of this document, a spray sunscreen product is one discharged from either a pressurized or nonpressurized container, with the understanding that the degree of atomization will likely vary according to the formulation, the container system used, and the design of the spray actuator, among other factors.
b. Spray sunscreen performance and effectiveness. The Dosage Form ANPR asked a series of questions relating to the performance and effectiveness of spray sunscreens, including questions about the amount of spray sunscreen typically applied by consumers, uniformity of coverage, how frequently consumers reapply spray sunscreens, whether consumers rub spray sunscreens into the skin when directed to do so and the resulting effect on effectiveness, and whether—and if so, how—the SPF and/or broad spectrum tests need to be modified to address sunscreen sprays. The Dosage Forms ANPR also solicited studies comparing spray sunscreens to other eligible dosage forms to see whether the dosage forms are comparable.
Four comments provided data from multiple studies examining and comparing the performance of spray and lotion sunscreens on a variety of parameters, including amounts applied, uniformity of coverage as measured with UV filter photography, comparative SPF results, and consumer ratings of ease and effectiveness of application, among others. FDA's evaluation of the information submitted indicated that key questions asked in the Dosage Forms ANPR were directly or indirectly addressed by these studies. These studies indicated that consumers like the convenience of spray sunscreens and adapt their use of these products to achieve effective coverage. Data provided on application uniformity lacked study reports and were difficult to compare directly, but—taken together—they suggest a high degree of uniformity between sprays and lotions in coverage of exposed skin, as well as between different spray application scenarios such as spraying directly on skin or spraying followed by rubbing. Information submitted indicated that the amount of spray sunscreen dispensed is higher than the amount dispensed for sunscreen lotions, and that consumers are more likely to reapply sprays than lotions. There was no response from any stakeholder regarding consumers' compliance with directions to rub a spray sunscreen into the skin. However, data was provided suggesting that rubbing spray sunscreens into the skin did not enhance effectiveness. Based on these comments and the available data, we are not proposing to require that labeling provide instructions to rub spray sunscreens into the skin.
Comments on the Dosage Forms ANPR also agreed, and we concur, that the current FDA-required SPF and broad spectrum tests are appropriate for evaluating the efficacy of sunscreens in spray dosage forms. SPF testing requires application of a set amount of sunscreen (2 mg/cm2 on each test subject), which can readily be done for spray sunscreen formulations. For example, comments on the Dosage Form ANPR stated that the SPF testing of sunscreen spray products can be conducted following the method described in the current rule by weighing out the liquid form and applying it to the skin. This premise is supported by data from SPF testing submitted in the comments. For example, one comment submitted five SPF testing reports conducted on sprays using the FDA-required methods, in which the expected SPF values for the test formulations were almost identical to the SPF testing results. The same logic applies to broad spectrum testing, which also uses a defined amount of sunscreen by weight. Based on this information, we conclude that the current and proposed SPF and broad spectrum testing methods are also appropriate for spray dosage forms.
c. Spray sunscreen safety. FDA has identified two primary safety concerns specific to spray sunscreen dosage forms: (1) The potential risk of respiratory harm from inhaling sunscreen ingredients and (2) the potential flammability risk when consumers are exposed to flame or heat before spray solvents have completely dried. For the reasons described below, we believe that both potential risks can be acceptably mitigated by proposed formulation limitations, labeling requirements, and adequate testing, and Start Printed Page 6231thus propose to establish these as additional conditions in the monograph to ensure that sunscreen products in a spray dosage form would be GRASE.
d. Inhalational toxicity. Broadly speaking, the human respiratory system consists of the upper respiratory tract (i.e., the airways of the nose to the larynx) and lower respiratory tract (the trachea and branching airways of the lung, including bronchi, bronchioles, and alveoli) (see generally Refs. 148 and 149). Much of the respiratory system is lined with a layer consisting of mucus cells and cilia that mechanically propel inhaled particles out of the lower respiratory tract toward the mouth, where they may be swallowed or expectorated (Refs. 148 and 149). The most significant concern associated with any product that may be accidentally inhaled is the potential risk of adverse effects associated with deep lung deposition, which occurs when particles in an aerosol (i.e., a suspension of airborne particles such as a sunscreen spray) reach the unciliated airways in the lung. Particles that can reach the unciliated airways of the deep lung are described as respirable and may be associated with serious adverse effects such as asthma, emphysema, bronchospasm, or chronic obstructive pulmonary disease; particles that do not reach the deep lung may be associated with less harmful adverse events such as local irritation of the upper airway, coughing, or sneezing (Refs. 149 to 151). The potential health risk associated with inhalation of hazardous aerosols depends on how much of a toxic substance is deposited in a given region of the respiratory tract and how much remains after physiological clearance occurs through mechanisms such as coughing, sneezing, mechanical transport, or, in the deep lung, engulfment by specialized cells or other protective action (Refs. 148 and 152).
The pathogenic potential of inhaled aerosols depends on where in the respiratory tract a particle is deposited (Ref. 152). Whether spray particles that enter the body through inhalation at the nose or mouth will be deposited in the lung depends largely on their physical characteristics: Most notably particle size, with the likelihood of respirability increasing as particle size decreases (Refs. 148 to 153). The effects of particle size on respirability of inhaled particles is well studied. There is general agreement that particles greater than 10 micrometers (μm) in diameter may enter the mouth and the airway up to the larynx. Approximately 50 percent of particles up to 10 μm in diameter can penetrate beyond the larynx to the thoracic region of the respiratory tract, while only particles smaller than 4 μm reach the unciliated airways and alveolar region of the lungs (see generally Refs. 148 to 153). Thus, although there are little or no data on the potential inhalation toxicity of particular spray sunscreen ingredients, we are proposing that exposure to harmful levels of such ingredients can effectively be minimized by imposing particle size limitations on spray sunscreen products.
Several comments on the Dosage Forms ANPR submitted results of particle size distribution testing using available methods and apparatus, with the aim of showing that exposure to inhaled sunscreen products or ingredients would be minimal and thus unlikely to cause adverse effects. The data submitted were similar and in some cases overlapping. In an analysis of pooled particle size distribution data from all submissions, representing 50 U.S.-marketed spray sunscreen products, 32 had particles smaller than 4 μm in diameter and thus within the respirable portion of the total particle size distribution. However, the great majority of the particle sizes observed were nonrespirable. The highest percentage that any product had of particles smaller than 4 μm in diameter was 0.43 percent and the mean was 0.22 percent, which is extremely low.
In addition to reviewing information from comments on the Dosage Forms ANPR, FDA conducted its own analysis of particle size distribution for 14 marketed spray sunscreens. In those tests, no sunscreen had more than 10 percent of particles in sizes less than 10 μm in diameter and only three had particles smaller than 5 μm (Ref. 154).
To limit the risks of unintentional exposure and potential associated adverse events to respirable particles in spray sunscreens, we are proposing limits on the size of particles dispensed from the consumer container for finished spray sunscreens in order for those products to be GRASE. We propose that 90 percent of the particles dispensed from the consumer container must be at least 10 μm or greater in order to limit exposure beyond the larynx, and to prevent deposition in the deep lung, the minimum particle size dispensed from the consumer container must be no less than 5 μm. This limit was chosen because it is the lowest whole number above the generally accepted threshold (4 μm) at which particles enter the unciliated airway and because it allows for experimental error that may be inherent in particle size measurements. Sunscreen products that do not meet both limitations would not be GRASE because there is not sufficient information in the record to support a positive finding about their safety. We believe that, taken together, these two limitations would significantly reduce inhalation risk from spray sunscreens by reducing particle exposure to the larynx and deeper lung tissues. The particle size data submitted in response to the Dosage Forms ANPR also suggest that these limitations would be readily achievable without unduly burdening sunscreen spray manufacturers.
With the establishment of these two limits, FDA believes that the risks of adverse events related to unintentional inhalation of spray sunscreens will be minimal. Stakeholders asserted that the risk of inhalation toxicity is already low, primarily based on particle size of marketed sprays. Limited data on adverse event reports and animal toxicity studies were also submitted in a few comments on the Dosage Forms ANPR, but were inadequate to support the safety of spray sunscreens in the absence of particle size limitations. If the particle size limitations proposed here are adopted, however, we do not believe that additional animal toxicity or other safety data need to be provided to support a GRASE finding for spray sunscreens.
We are proposing that particle size testing to demonstrate compliance with the proposed limitations must be conducted on spray products as they are dispensed from the consumer container as part of the lot release testing that would be routinely completed as part of current good manufacturing practice (CGMP) compliance under part 211 (21 CFR part 211). It is necessary to test the size of particles dispensed from the consumer container to ensure that particle size requirements are met under conditions of use by consumers.
For purposes of these proposed particle size requirements, we are using the term particle size broadly to mean the discrete unit emitted from the spray container that is available for inhalation by a consumer when the product is applied. If the particle dispensed from the consumer container is a droplet that meets the size requirements, the consumer will not accidentally inhale it into the deep lung. However, if that same droplet breaks apart into smaller fractions when it is dispensed from the consumer container, those fractions would be the particles that must meet the size requirement to ensure that consumers will not inadvertently inhale them past the larynx.
We are not proposing a specific test methodology for spray sunscreen particle size. Rather, sunscreen manufacturers would be obligated to ensure that particle size testing for their Start Printed Page 6232sunscreen sprays would be conducted on each lot of the final product as dispensed from the consumer container in accordance with adequate written specifications. USP General Chapter 601 part B provides methodology and requirements for sprays, aerosols, and powders that include methodology to determine droplet/particle size distribution, and we expect to consider testing done in accordance with the USP as adequate to meet this proposed requirement (Ref. 155).
We note that several comments on the Dosage Forms ANPR expressed concern about the potential inhalation risk from exposure to spray sunscreens that contain nanomaterials (as both active and inactive ingredients). One comment also recommended that FDA require the presence of such ingredients to be disclosed on spray sunscreen labels. FDA's approach to nanotechnology and nanomaterials in sunscreen products is discussed in section VII.E. FDA is not now proposing conditions of use, including labeling, for spray sunscreens that distinguish based on the presence of nanomaterials because we are proposing that any sunscreen spray that contains any particles smaller than 5 µm when it is dispensed from the consumer container would not be GRASE. With respect to nanomaterials in spray sunscreens, we note that the primary determinant of inhalation risk is the size of the particles in emitted sprays, which may be larger than individual formulation components. Nanoscale ingredients would not pass the particle size limitations for spray sunscreens; therefore, if they were to be detected when sprayed from the consumer container during particle size testing, the sunscreen could not be marketed under the OTC monograph.
In addition to the proposed limitations on particle size for sunscreen sprays and related testing, we are proposing to require that the following labeling be included in the directions for sunscreen sprays to minimize unintended inhalation:
- Hold container 4 to 6 inches from skin to apply.
- Do not spray directly into face. Spray on hands then apply to face.
- Do not apply in windy conditions.
- Use in a well-ventilated area and avoid inhalation.
This language is the same as that published in the Dosage Forms ANPR. Its adoption was supported by comments on the Dosage Forms ANPR, and the language is widely used on currently marketed spray sunscreens consistent with the 2018 Final Guidance.
e. Flammability risk. In July 2013, FDA issued a Consumer Update regarding persons catching on fire while wearing spray sunscreen products near an open flame:
The Food and Drug Administration (FDA) has become aware of five separate incidents in which people wearing sunscreen spray near sources of flame suffered significant burns that required medical treatment. The specific products reported to have been used in these cases were voluntarily recalled from the market, so should no longer be on store shelves. . . . In the five incidents reported to FDA, however, the burns occurred after the sunscreen spray had been applied. The ignition sources were varied and involved lighting a cigarette, standing too close to a lit citronella candle, approaching a grill, and in one case, doing some welding (Ref. 156).
These cases all involved a single manufacturer's product that has since been voluntarily recalled. Review of adverse event reports since the voluntary recall of this product indicates that no additional cases involving spray sunscreens have been reported. However, sunscreens are often used in very hot outdoor environments with high ambient air temperatures. Sunscreens are also frequently used around sources of flame or sparks, such as grills, bonfires, smoking, or other ignition sources. To ensure safe use of spray sunscreens and to better inform consumers about potential flammability risks, we are proposing to limit the flammability and require flammability labeling of spray sunscreens under the OTC sunscreen monograph.
FDA's general labeling regulations for OTC drugs provide for OTC monographs to require flammability labeling in suitable cases (§ 201.66(c)(5)(ii)(C)) (21 CFR 201.66(c)(5)(ii)(C))), and we have done so for products such as topical antitussives (21 CFR 341.74) and wart removers (21 CFR 358.150). As we did for those products, we are proposing to require each spray sunscreen formulation to be labeled for flammability in accordance with the testing methodology described in a regulatory provision issued by the Consumer Product Safety Commission (CPSC) (see 16 CFR 1500.43a). We have proposed to incorporate this flash point testing methodology to address our concern regarding the flammability of sunscreen in the spray dosage form after it has been dispensed onto the skin. We therefore propose that all batches of sunscreen spray products be tested for flammability in accordance with 16 CFR 1500.43a as part of batch release testing conducted in accordance with CGMP requirements.
We are also proposing to define three flammability categories for use in regulating and labeling sunscreens: (1) Extremely flammable, (2) flammable, and (3) combustible. These definitions refer to flash point testing to be performed using the method described in 16 CFR 1500.43a. These definitions are analogous to certain CPSC definitions located at 16 CFR 1500.3. Given the conditions under which sunscreens may be used, we are proposing that spray sunscreens found to meet the definition of extremely flammable in proposed § 352.3(f) are not GRASE and may not be marketed under the OTC sunscreen monograph. Products found to meet the definition of flammable or combustible in proposed § 352.3(g) or (h) would be required to include the following language in the “Warnings” section of the drug facts labeling: [bullet] “Flammable” or “Combustible” [as applicable] followed by a colon and the statement “Keep away from fire or flame”.
A further concern related to flammability is the time required for volatile solvents in a spray product to dry on the skin before a consumer can safely approach a source of heat or flame or can smoke without danger of fire. Typical sunscreen spray formulations contain 50 to 80 percent of a volatile carrier, most commonly ethyl alcohol. These volatile solvents are necessary to the formulation to allow the product to be sprayed onto the skin. After spraying, the solvents are intended to rapidly evaporate leaving a film of UV filters on the skin surface as the product dries. Once a spray product is dry, the solvent is no longer present so the flammability risk is low. However, prior to this point, the flammability risk would be higher.
We think that consumers should be warned to stay away from sources of flame while a flammable or combustible sunscreen spray dries. For this reason, we propose to require that each batch of a sunscreen spray product that meets the definition of flammable or combustible at § 352.3(g) or (h) be tested for drying time in accordance with written specifications. If the drying time is less than 5 minutes, we propose to require that the labeling state, “Wait 5 minutes after application before approaching a source of heat or flame, or before smoking.” If the drying time is at least 5 minutes but less than 10 minutes, we propose that the labeling would state, “Wait 10 minutes after application before approaching a source of heat or flame, or before smoking.” We propose that a sunscreen spray that is flammable or combustible and that takes 10 minutes or more to dry would not be GRASE because of the possibility of consumers approaching sources of fire Start Printed Page 6233during such an extended drying period. We invite comment on this approach.
4. Powder Dosage Forms
Although we have found powder sunscreens to be eligible for consideration in the OTC sunscreen rulemaking, we have tentatively determined that additional data as outlined below will be needed to support a conclusion that sunscreens in this dosage form are GRASE and to support consideration of appropriate labeling. Also, like sprays, powder sunscreens pose the potential for unintended inhalation, and for this reason, if admitted to the sunscreen monograph, the same limitations as to particle size here proposed for sprays would be expected to apply. For powder sunscreens that meet the particle size limitations proposed for sprays, we do not expect that additional toxicology data would be needed to address the potential health risks associated with inhalation.
One comment on the Dosage Forms ANPR provided data on SPF and broad spectrum performance of five powder formulations, as well as data from repeated insult patch tests and photosensitivity studies that were asserted not to show any safety issues. FDA has conducted particle distribution testing on five powder sunscreens. The powder sunscreens tested had a larger proportion of relatively small particles compared to the sprays. Only one of the five powder sunscreens would have complied with the requirement we are considering that no more than 10 percent of the particles could be smaller than 10 µm in diameter, and that product was also the only one that would have met the prospective limitation of no particles smaller than 5 µm in diameter (Refs. 153 and 154). Based on the data submitted, we believe that (current and proposed) SPF and broad spectrum test methods are appropriate for use with powder sunscreens, and we are not requesting additional respiratory safety information for powders that meet the same particle size limitations proposed for spray sunscreens.
FDA invites comments and data on the following topics related to powder sunscreens:
- What amounts of powder sunscreens do consumers typically dispense?
- What amounts of powder sunscreens are effectively transferred to the skin?
- How uniform is the sunscreen application across the sun-exposed area of the skin?
- How frequently do consumers reapply the product?
- Does rubbing a powder into the skin change sunscreen effectiveness?
- Are powder dosage forms water-resistant? If they are not water-resistant, is a direction to reapply every 2 hours sufficient to assure their safe and effective use?
- Can the powder dosage form be used safely and effectively over all areas of skin exposed to the sun, or should this dosage form be limited to the face?
- What factors, if any, should FDA consider in connection with particle size limitations or test methods for sunscreen powders?
- Are there important differences among powder types (e.g., loose, compact) or applicators that would affect particle size testing?
FDA will evaluate data and information submitted in response to these questions, as well as any other submitted or available data, to determine whether additional data are needed to support a final GRASE determination for this dosage form.
B. Proposed Maximum SPF and Broad Spectrum Requirements
In the Stayed 1999 Final Monograph, FDA established SPF 30+ as the maximum labeled SPF value for sunscreen monograph products, and required that each sunscreen monograph active ingredient contribute a minimum SPF of 2 to finished sunscreen products (64 FR 27666 at 27672, 27674 and 27675). The final monograph did not include any broad spectrum protection provisions. In its 2001 decision to stay the final monograph, however, FDA indicated that it was issuing the stay because the Agency intended to amend the sunscreen monograph to address requirements for both UVA and UVB radiation protection (66 FR 67485). FDA later addressed these issues in the 2011 L&E Final Rule, which, among other things: (1) Established optional broad spectrum labeling based on satisfaction of a critical wavelength test, (2) created an optional indication relating to skin cancer and early skin aging risk reduction for broad spectrum products with an SPF of 15 or higher, and (3) required a labeling warning for sunscreens that did not both satisfy the broad spectrum test and provide an SPF of at least 15 (76 FR 35620 at 35626-35628) (L&E Final Rule). Concurrently with publication of the L&E Final Rule, FDA issued a proposed rule to raise the maximum labeled SPF value for sunscreen products containing sunscreen monograph active ingredients to SPF 50+ (76 FR 35672, June 17, 2011).
In the time since these 2011 publications, the body of evidence in the published literature on UVA radiation (particularly UVA I radiation) and its role in the development of skin cancer has grown. This new data about the harms of UVA exposure is a significant concern given, among other things, that with currently available sunscreens, consumers may unknowingly accumulate excessively large UVA doses by using sunscreens with high SPF values that either: (1) Do not pass FDA's current critical wavelength-based broad spectrum test or (2) have inadequate uniformity in their UVA protection. Because of these concerns, we are making a number of proposals designed, among other things, to couple a greater magnitude of UVA protection to increases in SPF values.
1. Background
UV radiation includes both UVA and UVB rays. UVB rays (i.e., those with wavelengths from 290 to 320 nm) are higher energy, are much more effective at producing sunburn, and produce greater amounts of cellular damage (including DNA lesions, which can result in gene mutations linked to skin cancers) (Refs. 157 and 158). UVA rays (i.e., those with wavelengths from 320 to 400 nm) are lower energy and less effective at producing sunburn, but make up the majority of UV radiation, and penetrate much deeper into the skin, potentially causing oxidative damage (through formation of ROS) to skin pigment cells (Ref. 159). UVA rays also contribute to photo-aging (Ref. 157 and 160). Although the current scientific literature attributes UV-signature DNA lesions primarily to UVB wavelengths, UVA wavelengths can also produce DNA lesions. Although UVA wavelengths produce DNA lesions to a significantly lesser degree than UVB wavelengths do, DNA lesions produced by UVA rays have been reported to have slower repair rates (Ref. 157). UVA rays are comprised of UVA I rays (340 to 400 nm) and UVA II rays (320 to 340 nm). As discussed below, until recently, UVA I rays were generally not considered to contribute significantly to the harms associated with UV exposure.
Sunscreen products must be labeled with an SPF value calculated using a standardized SPF testing procedure set forth in FDA regulations (see § 201.327(i)). ISO 17166 CIE S 007/E was approved for incorporation by reference into § 201.327(i) as of June 18, 2012 (76 FR 35619, June 17, 2011). The SPF test measures the amount of UV radiation exposure it takes to cause Start Printed Page 6234sunburn when a person is using a sunscreen compared with how much UV exposure it takes to cause sunburn when the person is not using a sunscreen. Sunscreens with increasing SPF values (up to a certain point) have been demonstrated to provide increased sunburn protection. Because SPF values represent a sunscreen's level of sunburn protection, they are primarily (though not exclusively) an indicator of expected protection from UVB radiation. To pass FDA's current test for broad spectrum labeling (§ 201.327(j)), however, sunscreens must demonstrate that, in addition to UVB protection, they also provide some UVA protection.
Only products that have been determined to have a minimum SPF value of 15 and to pass our broad spectrum test may include statements in their labeling indicating that they decrease the risk of skin cancer and early skin aging caused by the sun when used as directed with other sun protection measures (§ 201.327(c)(2)). In contrast, sunscreens that have not been determined to provide both broad spectrum protection and an SPF value of at least 15 must include a skin cancer/skin aging alert warning to consumers that “[s]pending time in the sun increases your risk of skin cancer and early skin aging” and that “[t]his product has been shown only to help prevent sunburn, not skin cancer or early skin aging” (§ 201.327(d)(2)).
2. Increased Evidence of Harms Associated With Exposure to UVA Radiation
Since publication of the 2011 L&E Final Rule and Max SPF PR, the strength of scientific evidence linking UVA exposure to skin cancers and other harms has increased. This evidence suggests that UVA wavelengths continue generating DNA lesions hours after UV exposure (Ref. 161) and that if left unrepaired, these DNA lesions can form UV-induced mutations in many genes that have been detected in both melanoma and nonmelanoma skin cancers (Refs. 162 to 165). Further, unlike UVB-induced DNA lesions, which attenuate with skin depth, recent evidence indicates that DNA lesions induced by UVA I exposure show the opposite pattern, with both increased DNA lesions in the basal layer of the epidermis (where melanocytes and proliferating keratinocytes reside) and less efficient DNA lesion repair in the basal layer (Refs. 166 and 167).
Damage to cells in the basal layer (if left unrepaired or if inefficiently repaired) can lead to mutations in critical genes that increase the possibility that normal cells will transform into cancer cells. While inefficient DNA repair is a concern for all individuals exposed to UV radiation, this concern is particularly acute in those with xeroderma pigmentosum (a disease caused by a disorder of the DNA repair system), who have extreme sensitivity to UV radiation, and who develop both nonmelanoma skin cancer and melanoma with a high frequency and very early in life (Ref. 168). In addition to the skin cancer-related risks associated with UVA exposure, increasing evidence shows that UVA I radiation also produces immunosuppression (Refs. 169 and 170). This, too, is a general concern for all individuals, but is especially dangerous for certain at-risk populations (such as organ transplant recipients and others on immunosuppressive drugs).
Given the above-described evidence, we are concerned about the existing potential for inadequate UVA protection in marketed sunscreen products. This is a particular concern with respect to high SPF sunscreen products that do not pass FDA's current critical wavelength-based broad spectrum test or that (though they pass our current broad spectrum test) have inadequate uniformity in their UVA protection. Consumers using these products may, while successfully preventing sunburn, accumulate excessively large doses of UVA radiation, thereby exposing themselves to additional risks related to skin cancer and early skin aging. The International Agency for Research on Cancer has found that high SPF sunscreen products are associated with longer intentional UV exposures (Ref. 171), raising the concern that use of these products may result in significant doses of UVA radiation. We note that concerns relating to inadequate UVA protection came up in several comments we received in response to the 2011 Max SPF PR, and that these comments raised particular concerns about inadequate UVA protection in high SPF products. This concern has also grown over time in the published literature (Refs. 172 to 175).
For all of these reasons, we are proposing a number of steps designed to couple a greater magnitude of UVA protection to increases in SPF values. As discussed in further detail below, we are also making proposals designed to address evidence of variability in SPF values and evidence showing additional clinical benefits associated with SPF 60 sunscreens.
3. Broad Spectrum Proposals
a. UVA I/UV ratio required to pass the broad spectrum test. We are proposing certain changes to the requirements to pass the broad spectrum test. Specifically, we are proposing to add to the current broad spectrum test a requirement that products meet a UVA I/UV ratio of 0.7 or higher. We note that the current broad spectrum test procedure would remain unchanged [40] and that this new ratio would be calculated using data from the existing test, which should help minimize burden on manufacturers.
The current labeling regulation requires that sunscreens labeled as broad spectrum achieve a critical wavelength of 370 nm or greater (§ 201.327(j)). A sunscreen product's UV protection is often displayed as a curve on a graph showing the amount of UV absorbance the product provides at each wavelength in the UV spectrum (i.e., from 290 to 400 nm). The “critical wavelength” of the product is the wavelength corresponding to 90 percent of the area under this curve. Higher critical wavelengths, therefore, illustrate greater breadths of UV protection across the 290 to 400 nm spectrum.
Most sunscreen products—even if they achieve a critical wavelength of 370 nm or greater and therefore meet the current criteria for broad spectrum labeling—have historically covered the UVB and UVA II ranges preferentially. Given how much of the UVA portion of the UV spectrum is composed of UVA I radiation (see Figure 3 below) and given what we now know about the risks associated with UVA exposure, and with UVA I exposure in particular, ensuring that sunscreen products provide adequate protection in the UVA I portion of the spectrum is critical.
Start Printed Page 6235
We are therefore proposing to require that in order to pass the broad spectrum test, a product must demonstrate that it provides a UVA I/UV ratio of 0.7 or higher, indicating that the product provides a minimum measure of UVA I radiation absorbance relative to total UV radiation (i.e., UVB + UVA) absorbance, in addition satisfying to the 370 nm critical wavelength requirement. Requiring a UVA I/UV ratio of 0.7 or higher for broad spectrum products would mean that these products would have a more uniform amount of radiation protection across the UVA I, UVA II, and UVB ranges. This improved fidelity across the UV spectrum is especially important for high SPF products which, as discussed above, are associated with longer intentional sun exposure, which in turn can result in significant doses of UVA radiation. This proposed UVA I/UV ratio would also help eliminate the current potential for a product labeled as broad spectrum that has a higher SPF value to provide (unbeknownst to the consumer) poorer broad spectrum protection than a product labeled as broad spectrum with a lower SPF value (depending on the particular combination of active ingredients used in the product and which parts of the UV spectrum they absorb). For example, under the current testing regime, a sunscreen that is labeled “broad spectrum SPF 30” could provide less UVA protection than a sunscreen labeled “broad spectrum SPF 15.”
We note that FDA first raised concerns relating to the adequacy of UVA protection in sunscreen products in 2007 (see 72 FR 49070 at 49104 to 49107). At that time, we proposed a similar ratio to the one we are proposing today as part of a different, more complex proposal for testing and labeling to address broad spectrum protection that, among other things, included both in vitro (spectrophotometric) and in vivo (clinical) testing for UVA radiation, as well as a four-tier UVA star rating labeling system. In response to comments describing purported disadvantages of that proposal, including general comments that the proposal was overcomplicated, specific comments on the proposed in vitro testing method, and comments indicating that “[t]he proposed ratio places too much emphasis on the UVA I region, which is not generally considered to contribute significantly to the harmful effects of exposure to UV radiation” (76 FR 35620 at 35650), we made a number of changes to our 2007 proposal in the 2011 L&E Final Rule. Those changes included elimination of the UVA I/UV ratio and adoption of the above-described critical wavelength test for establishing broad spectrum protection instead. As we noted in the preamble to the L&E Final Rule, our decision not to require the UVA I/UV ratio at that time was based, in part, on our agreement with comments stating that the scientific evidence available at that time indicated that UVA I exposure did not pose sufficient risk of harm to justify the emphasis placed on it by the ratio, and that the critical wavelength test provided a superior measure of broad spectrum protection (id. at 35650).
As described above, in the time since issuance of the L&E Final Rule, the body of evidence showing the harms of UVA exposure, and of UVA I exposure, in particular, has grown significantly (Refs. Start Printed Page 6236159, 161, 162, 166, 167, and 169). It is now clear that in addition to producing the immunosuppression described above, UVA I exposure also results in increasing DNA damage with increasing skin depth (in contrast to UVB-induced DNA damage, which is reduced as skin depth increases). In addition, given that UVA I is the predominant portion of UVA radiation, new evidence (discussed in section IX.B.2) strengthening the link between UVA radiation and skin cancer development raises our concerns about the potential for inadequate protection in the UVA I portion of the UV spectrum. Accordingly, we no longer agree with the earlier comments suggesting that UVA I does not contribute significantly to the harmful effects of exposure to UV radiation, or with our 2011 conclusion that a UVA I/UV ratio requirement would therefore place too much emphasis on this portion of the UV spectrum.
We emphasize that we are not proposing to replace the existing critical wavelength test, and that the proposed ratio would supplement (and be calculated using data from) the existing broad spectrum test. We also note that the UVA I/UV ratio we are proposing would result in a level of UVA protection similar to what is achieved via the European Union's recommended minimum UVA protection factor of 1/3 of the labeled SPF and via the United Kingdom's Boots 3-star rating (the United Kingdom has for decades used a tiered star rating system based on an alternative ratio method) (Refs. 173 and 174). We note that data collected in 2009 about 330 sunscreen products commercially available in the United States showed that, at that time, more than half of these products already satisfied the broad spectrum test we are now proposing (see Comment, Docket No. FDA-1978-N-0018-0690).
b. Broad spectrum requirement for all products that are ≥SPF 15. We are also proposing to require that all sunscreen products with SPF values of 15 and above demonstrate that they provide more uniform protection across the UVA I, UVA II, and UVB ranges of the UV spectrum by satisfying FDA's revised broad spectrum test. This proposal is designed to link increases in SPF value not only to increases in UVB protection, but to increases in the magnitude of UVA protection as well. We note that a consumer using a sunscreen that provides robust protection against sunburn but that does not pass FDA's revised broad spectrum test—and therefore provides inadequate UVA protection—may fail to get out of the sun, thereby exposing themselves to higher levels of UVA radiation than if they had not been protected from sunburn. Given the increasing evidence of major health risks associated with UVA exposure, we propose to find that such products (those with SPF values of 15 and greater that do not provide sufficient protection across the UV spectrum (as demonstrated by satisfying FDA's revised broad spectrum requirement)) are not GRASE. At the same time, we conclude that the evidence described above regarding the contribution of UVA I to skin carcinogenesis, coupled with the evidence reviewed in the 2011 L&E Final Rule (see 76 FR 35620 at 35630-35634), supports the proposal to include sunscreen products that have an SPF of 15 or higher and also pass the revised broad spectrum test in the sunscreen monograph with indications both for use to help prevent sunburn and for use, as directed with other sun protection measures, to reduce the risk of skin cancer and early skin aging caused by the sun. As we indicated in the L&E Final Rule, the whole range of UV radiation, and not specific wavelengths, is a human carcinogen, and the exact wavelengths most responsible for these harmful effects are not known (see id. at 35631, 35633). To assure that a clinically meaningful reduction in the risks of skin cancer and early skin aging is achieved, then, a product must contribute to substantially limiting overall UVB and UVA exposure (see id. at 35630, 35631-35632), as will be assured by our proposal to couple the enhanced breadth of protection across the UVA spectrum provided by the revised pass criteria for the broad spectrum test with the magnitude of protection assured by requiring a minimum SPF of 15.
By requiring that all sunscreens with SPF values of 15 or more satisfy the (new) broad spectrum standard (including the new ratio requiring proportionate protection), this proposal will also enable consumers to select a product primarily by numerical (SPF) value on the label, having assurance that, when used as directed, a product labeled with a higher numerical SPF value provides proportionately more protection not only against sunburn, but also against skin cancer and skin aging than lower numbered products [41] (provided that the product provides an SPF of at least 15). In doing so, this proposal also eliminates another source of potential confusion permitted by the current labeling regime, in which a higher numbered product (for example, one labeled SPF 30) may provide inferior protection against UVA radiation than a lower numbered product (for example, one labeled Broad Spectrum SPF 15).
c. Sunscreen products with SPFs <15. As noted above and in section III.A.2, sunscreen products with SPF values below 15 have not been shown to reduce the risk of skin cancer or early skin aging caused by the sun, whether or not they provide broad spectrum protection. Because of this limitation, we considered proposing to remove from the monograph sunscreen products with SPF values lower than 15. However, as the Surgeon General has acknowledged (Ref. 5), some consumers may seek intentional sun exposure because (for example) they associate tanned skin with attractiveness and health. These consumers may seek some protection from sunburns and therefore, select a low SPF product (i.e., one with an SPF value below 15). If such products are removed from the market, these consumers may choose not to use a sunscreen product at all rather than use a broad spectrum product with an SPF of 15 or above.
Although the benefits of sunscreen products with SPFs below 15 (which are not indicated to reduce the risk of skin cancer or early skin aging) are limited, FDA believes that the use of such products is preferable to the use of no sunscreen at all. Thus, to provide sunburn protection for these consumers, FDA is proposing that sunscreens with SPF 2 to 14 that bear prominent labeling regarding their limited use for sunburn prevention and the risks associated with spending time in the sun (see sections IX.B.1 and IX.C) may remain on the market without approved NDAs. Because products with SPFs below 15 have not been demonstrated to reduce the risk of skin cancer, FDA is not proposing to require products with SPF values under 15 to pass the broad spectrum test. However, we seek comment on whether the limited benefits such sunscreen products confer outweigh the risks of sunscreen drug exposure and the potential false sense of security provided regarding UV protection (i.e., whether such sunburn-only sunscreen products are GRASE and should remain on the market without approved NDAs).
4. Maximum SPF Value Proposals
a. Maximum labeled SPF value would be SPF 60+. In conjunction with the broad spectrum proposals described above, we are also proposing to raise the maximum labeled SPF value for Start Printed Page 6237products containing sunscreen monograph active ingredients to SPF 60+. Under this proposal, sunscreen products with SPF values of 60 or greater would be labeled “SPF 60+.”
FDA has proposed to raise the maximum SPF value that sunscreens marketed pursuant to the OTC Monograph System can display on their labeling several times. In the 1978 notice of proposed rulemaking, we proposed that such sunscreens be labeled with a maximum SPF value of 15 (43 FR 38206 at 38213 to 38214). In the 1999 final monograph, we determined that that cap should be increased to SPF 30+ (64 FR 27666 at 27675). In 2007 (72 FR 49070 at 49085 to 49087) and then in 2011 (Max SPF PR), we tentatively concluded that data existed to show that sunscreens with labeled SPF values of up to 50+ provide additional clinical benefit to consumers. Our proposal today to increase the maximum labeled SPF value to 60+ is similarly based on data showing the additional clinical benefit provided by SPF 60 sunscreen products when those products also provide broad spectrum protection.
In the 2011 Max SPF PR proposing an SPF 50+ cap, we noted that the record, at that time, lacked adequate data demonstrating that sunscreen products with SPF values above 50 provided additional meaningful clinical benefit over and above what was provided by SPF 50 protection (76 FR 35672 at 35672 to 35674). We requested data showing that such clinical benefits existed (id.). In response to both the 2007 and 2011 proposals, we received comments providing citations to data showing the additional meaningful clinical benefit provided by sunscreen products with SPF values of 60 for certain at-risk populations when those sunscreens also included broad spectrum protection. (See, e.g., Ulrich et al. (showing statistically significant protection of organ transplant recipients, who are highly susceptible to nonmelanoma skin cancer, from squamous cell carcinoma with use of broad spectrum SPF 60 sunscreen) (Ref. 176); see also Comment FDA-1978-N-0018-0710, August 31, 2011, citing Kuhn et al. (showing statistically significant prevention of skin lesions in topical lupus erythematosus patients with use of broad spectrum SPF 60 sunscreen after exposure to either UVA I source or UVA II/UVB source) (Ref. 177); Faurschou et al. (showing prevention of urticarial reaction in subjects with idiopathic solar urticaria with use of broad spectrum SPF 60 sunscreen) (Ref. 178); Fourtanier et al. (showing lower levels of polymorphous light eruption in subjects using broad spectrum SPF 60 versus SPF 50 products (Ref. 179)). Based on the additional meaningful clinical benefit provided by broad spectrum SPF 60 sunscreens shown in these studies, we are proposing to raise the maximum labeled SPF value to SPF 60+.
Because the studies demonstrating the additional meaningful clinical benefit provided by SPF 60 sunscreens all used sunscreens that also provided broad spectrum protection, however, the additional clinical benefit shown to exist at SPF 60 cannot be decoupled from the broad spectrum protection provided by those products. That is, the additional meaningful clinical benefit shown in these studies may have been the result of the sunscreens' protection against rays in the UVB range or in the UVA range, or both. For this reason, our proposal to recognize the additional meaningful clinical benefit provided by sunscreens with SPF values above 50 is consistent with, and dependent upon, our proposal that all sunscreen products with SPF values of 15 and above be required to provide broad spectrum protection.
Given the lack of data showing that sunscreens with SPF values above 60 provide additional meaningful clinical benefit, however, we are proposing not to allow labeled SPF values higher than 60+. Labeling sunscreen products with SPF values higher than what has been shown to provide additional meaningful clinical benefit could have unintended negative consequences. For example, as discussed above, such products may inadvertently promote extended solar exposures because consumers feel protected and assume that the higher SPF value implies that greater UV exposure is safe (see, e.g., Autier, et al., 2007 (Ref. 171)).
b. Formulation cap for sunscreen products of SPF 80. Although we are proposing that the maximum labeled SPF value will be SPF 60+, we are proposing to permit the marketing of sunscreen products formulated with determined [42] SPF values up to 80. We are proposing to permit this additional formulation margin in part because of the inherent variability in SPF test results. A sunscreen product's SPF value is calculated from measurements that are based on an investigator's visual evaluation of an individual test subject's erythema response to a series of UV doses administered in successive sites on the subject's back. Because the administered UV dose series for the final minimal erythema dose (MED) [43] of a sunscreen with an expected SPF of 60 increases by 15 percent with each successive dose (see § 201.327(i)(5)(iii)), a difference in judgment of one site in opposing directions would result in up to approximately 30 percent variability in the assessment of the amount of exposure that resulted in the erythema.[44]
Allowing the marketing of sunscreen monograph products with determined SPF test results up to 80 would, therefore, more fully account for the range of variability in SPF test results for sunscreen products labeled SPF 60+. We are also proposing this formulation margin to provide manufacturers with additional formulation flexibility that we hope will help facilitate the development of products with greater UVA protection, given our expectation that active ingredients added for the primary purpose of increasing UVA protection would contribute to a sunscreen's determined SPF value as well. We seek comment on whether SPF 80 is the appropriate formulation cap to accomplish these objectives.
We are proposing not to allow the marketing (without an approved NDA) of sunscreen products with determined SPF values above SPF 80. This proposal follows from the principle that if the addition of ingredients to a drug does not provide additional clinical benefit but potentially increases the risk associated with the drug, this shifts the benefit-risk calculation and renders the drug not GRASE (see, e.g., 76 FR 35673 at 35675). In light of this principle, we solicited comments in 2011 on the appropriateness of a formulation cap for sunscreen products (id.).
Some of the comments that we received in response to the 2011 Max SPF PR expressed concerns (in general) about the safety of unnecessary exposure to sunscreen active ingredients. We received only one comment, however, directly addressing the question of an SPF formulation cap. That comment emphasized that there was no formulation limit in other countries using an SPF labeling cap, and Start Printed Page 6238that the list of permitted active ingredients in the monograph itself establishes an SPF ceiling for the formulation as a whole. FDA rejects the premise that the list of permitted active ingredients establishes an adequate SPF cap for sunscreen formulations, as this theory does not take into account the potential addition of new GRASE ingredients to the list of active ingredients under the monograph. This comment also appears to imply that the maximum concentration of each active ingredient correlates specifically to a particular numerical contribution to the total SPF value of the product. This has not been established (see 64 FR 27666 at 27674 and 27675 (noting that formulation techniques may enable increases in SPF without use of higher concentrations of active ingredients)). In addition, as mentioned in 2011 in the Max SPF PR (76 FR 35672 at 35674), the theoretical increase in protection implied by higher SPF values generated in a laboratory does not necessarily correspond to meaningful additional sunburn protection for consumers in actual use conditions. Given that a solar simulator in a lab can produce much higher UV doses than a consumer would receive from the sun (even in the most extreme situations), it is unlikely that a consumer could ever actually reach the theoretical ceiling created by the list of permitted active ingredients.
Given the lack of demonstrated clinical benefit for sunscreens with determined SPF values above SPF 60, and the potential for risks—discussed elsewhere in this document—associated with exposure to sunscreen active ingredients, we propose not to permit the marketing (without an approved NDA) of sunscreen products with determined SPF values above SPF 80 (which reflects a formulation margin intended both to give full effect to the SPF 60 limit and to enable formulation flexibility).
c. Proposal for ≥SPF 15 labeling. Finally, we are proposing to require that sunscreen monograph products with determined SPF values of 15 or above be labeled with an SPF number corresponding to the lowest number in a range of tested SPF results, as shown in table 5.[45] For example, sunscreens testing at SPF 15 to 19 would be labeled “SPF 15”; those testing at 40 to 49 would be labeled “SPF 40.” [46]
This proposal is designed to avoid misleading consumers about the relative efficacy of sunscreen products, given the lack of clinical data showing meaningful efficacy differences between closely grouped SPF values. We note that in the 2011 L&E Final Rule, FDA declined a request that SPF be labeled in multiples of five, stating that there was no mathematical or statistical basis for this labeling approach because SPF values could generally be determined with a precision that allowed for SPF values to be labeled in intervals of less than five units. New data showing variability both between tested SPF values for individual study subjects and for determined SPF results achieved across multiple labs testing the same sunscreen formulation (i.e., variability inherent in a clinical test that relies on visual assessments) (FDA-1978-N-0018-0740, 2011; Ref. 182), however, has caused us to reexamine this issue.
As described above, the clinical SPF test is conducted using a solar simulator to administer several specified doses of UV radiation that increase by 15 to 25 percent with each successive dose to a human subject's back in both sunscreen-treated and untreated areas (with the specific UV doses being derived from the expected SPF of the product and a determination of the individual subject's UV sensitivity). The clinical investigator then visually evaluates both the sunscreen-treated and untreated areas of the subject's back to identify the areas with perceptible skin redness (erythema) that has clearly defined borders. Determining which of several areas on a single subject's back should be considered to meet this “clearly defined borders” criteria is an exercise of clinical judgment. Once the investigator has made this judgment, he or she then records the smallest dose of UV radiation it took to create an area with the observed skin reaction of erythema with clearly defined borders. After assessing multiple individual test subjects this way, the resulting UV exposure information is used in calculating the determined SPF value of the sunscreen being tested. The data we reviewed suggest that the clinical evaluation undertaken during this process creates variability that justifies the use of SPF ranges.
For example, in a study using panels of five subjects, the mean SPF values observed across multiple labs ranged from 54 to 82 for a target SPF 80 (FDA-1978-N-0018-0740, 2011). This same study also evaluated a scenario where a lab was not told the target SPF, but was rather given a range of SPF 20 to 100 for a product with an expected SPF of 100. The results showed that it was extremely difficult for labs to reproduce the labeled SPF 100, with mean SPF values ranging from 37 to 75. In a second study with multiple panels of 25 subjects that was controlled and randomized, the determined SPF of two sunscreen formulations tested across four labs ranged from 63 to 69 for a target SPF 70 and from 82 to 89 for a target SPF 90 (Ref. 182). Although the magnitude of the differences observed in this second study were not statistically significant, the fact that multiple labs determined different specific numerical values for a single formulation suggests that the use of labeled values representing ranges more accurately represents the sun protection provided by a product, and therefore is appropriate to avoid misleading consumers.
We note that variability in SPF values is exacerbated at high SPFs. For example, individual test results with 30 percent variability from a determined SPF value of 20 would range from SPF 14 to SPF 26; individual test results with 30 percent variability from a determined SPF value of 50 would range from SPF 35 to SPF 65. Accordingly, as shown in table 5, we propose that the range of tested values reflected in the labeled SPF number should be wider at higher SPF values and narrower at lower ones, and that the requirement that labeled SPF values correspond to ranges rather than precise numerical values is not necessary below SPF 15.
Table 5—Proposed SPF Labeling Ranges
Range of determined SPF values Associated labeled SPF value 60-80 60+. 50-59 50. 40-49 40. 30-39 30. 25-29 25. 20-24 20. 15-19 15. 2-14 Determined SPF Value. C. Proposed PDP Labeling Requirements
We are also proposing some revisions to the principal display panel (PDP) for sunscreen products (the PDP is the portion of an OTC drug product label that is most evident when the product is displayed for retail sale (§ 201.60)). In addition to satisfying general OTC drug labeling requirements found in part 201, Start Printed Page 6239sunscreen product PDPs are currently required to satisfy specific labeling requirements in § 201.327. We are proposing to amend these requirements for sunscreen PDP labeling (currently codified in § 201.327(a) and (b), and (for the statement of identity of products that also include skin protectants) in § 201.327(h)) to help consumers better understand, evaluate, and compare sunscreen products by providing additional information on the PDP, and by ensuring the prominence and readability of information required to appear on the front of the container or package. We are also proposing to renumber and consolidate provisions on PDP labeling and the statement of identity (SOI) in § 201.327(b) to incorporate new proposed provisions in § 201.327(a), as described in section IX.D.2.b of this preamble. In addition, we are proposing that labeling a sunscreen product in accordance with proposed § 201.327(b) would be a condition for marketing a sunscreen under the OTC sunscreen monograph in part 352.
We are proposing to revise the current SOI, which is required to appear on the PDP by both current and proposed § 201.327. Currently, the SOI for sunscreens under this regulation contains “the established name of the drug, if any” and identifies the product as a “sunscreen.” The revised SOI would consist of an alphabetical listing of all sunscreen active ingredients in the product using the names shown in § 201.327, followed by “Sunscreen” and the product's dosage form (such as lotion or spray). In light of these proposed changes to the SOI for sunscreens, we are also proposing harmonizing changes to the provisions that address the SOI for products that combine sunscreen and skin protectant active ingredients (proposed § 201.327(h) and cross-referenced in the sunscreen monograph in § 352.60 (21 CFR 352.60) and in the skin protectant monograph in § 347.60 (21 CFR 347.60)).
The proposal to list all active ingredients as part of the SOI is generally consistent with SOI labeling of other OTC and prescription drugs. Providing information about a product's active ingredients and dosage form would supplement other important elements of the PDP (SPF, broad spectrum, and water resistance information) to provide a succinct summary of the product's key characteristics on the front of the package or container. We expect that this approach would enable consumers to more readily compare differing products and either select or avoid a given product accordingly. As an indication that consumers value information about a sunscreen's active ingredients, an analysis of top-rated sunscreen product reviews on Amazon.com found that product ingredients were listed as a positive factor in 17 percent of responses, and a negative factor in 10 percent of responses (Ref. 183).
Based on a review of marketed sunscreen product labels, FDA is concerned that the SOI may currently be obscured by the inclusion and prominence of other printed or graphic information on the PDP. For this reason, we also propose to require the SOI to appear in direct conjunction with the most prominent display of the proprietary name, in a boldface font at least one-fourth the size of the most prominent printed matter on the PDP, and displayed so that the text is generally parallel to the base of the packaging. We propose that the entire SOI appear in the same font style, size, and color with the same background color, and as continuous text with no intervening text or graphic material other than text provided in accordance with the requirements for the SOI for a product that also includes a skin protectant, where applicable. These requirements would supplement, and not replace, the general requirements regarding the PDP and SOI for all nonprescription products in §§ 201.60 and 201.61.
Proposed § 201.327(b) would incorporate the “Broad Spectrum SPF,” “SPF,” and “Water Resistant” statements that already must appear on the PDP as described in current § 201.327(a). Additionally, for all products with SPF values below 15, we propose to require that the SPF statement be followed by an asterisk (*) directing the consumer to the statement “*See Skin Cancer/Skin Aging Alert.” We propose that the quoted statement must appear in the bottom 30 percent of the PDP. This statement is intended to draw the consumer's attention to the Skin Cancer/Skin Aging Alert that would continue to be required for these products as part of the “Warnings” in the Drug Facts portion of the label (§ 301.327(d)), because there is evidence that some sunscreen consumers are not reading this information in its current location (Refs. 184 and 185).
Under the current regulation, the entirety of the “Broad Spectrum SPF” or “SPF” statement, as applicable, must appear on the sunscreen PDP in the same font style, size, and color and with the same background color, and, if used, the “Broad Spectrum SPF” statement must also appear as continuous text with no intervening text or graphic. To further ensure the prominence and readability of information that is important for consumers to evaluate and compare sunscreen products, we propose that these statements must also appear in bold typeface at least one-fourth the size of the most prominent printed matter on the PDP, and as text generally parallel to the base of the packaging.
The proposed new “*See Skin Cancer/Skin Aging Alert” statement would also be required to appear in bold typeface at least one-fourth the size of the most prominent printed matter on the PDP, and as text generally parallel to the base of the packaging. In addition, the entire statement would appear in the same font style, size, and color with the same background color, and as continuous text with no intervening text or graphic.
Finally, because water resistance is also an important characteristic for consumers when choosing a sunscreen, we also propose to apply comparable format requirements to the current “Water Resistant” statement. The statement would also be required to appear in bold typeface at least one-fourth the size of the most prominent printed matter on the PDP, and displayed so that the text is generally parallel to the base of the packaging. In addition, the entire statement would appear in the same font style, size, and color with the same background color, and as continuous text with no intervening text or graphic.
D. Proposed Requirements Related to Final Formulation Testing and Recordkeeping
We are also proposing a number of revisions in § 201.327: (1) To ensure that efficacy testing of the sunscreen formulation to be marketed is conducted in a way that protects human subjects and produces reliable results and (2) to enable FDA to assess compliance with this section's provisions going forward. We also propose to make compliance with these requirements a monograph condition in part 352.
1. General Approach to Final Formulation Testing
Current § 201.327 includes technical instructions for conducting the final formulation testing required to support the SPF values, water resistance statements, and broad spectrum statements shown in sunscreen product labeling. However, the current regulation does not explicitly address important broader considerations that are essential to ensure that final formulation testing is conducted and Start Printed Page 6240documented in a way that verifiably provides for protection of human subjects in SPF and water resistance testing, as well as ensuring the reliability of all the testing data that underlies sunscreen labeling. We expect that persons responsible for conducting final formulation testing should already be following best practices in their current testing programs. However, we are concerned that many entities may not uniformly observe such practices and/or may not maintain the records needed to document compliance with the final formulation testing procedures set forth in § 201.327. FDA's experience in conducting inspections and other actions to verify testing under the current provisions of § 201.327 have suggested latent problems in these areas. Although limited, this experience reinforces FDA's belief that further clarification of regulatory expectations is necessary given the public health importance of ensuring that sunscreen products are effective and accurately labeled, and the broad range of entities that may be involved in bringing sunscreen products to market. Thus, we are proposing to incorporate FDA's current expectations more explicitly in the revised provisions. The proposed provisions are broadly consistent with current best practices for efficacy testing conducted in human subjects, and are not expected to require significant changes by reputable and experienced testing establishments. Key areas of concern that are addressed by the proposed revisions include the following.
a. Protection of human subjects and oversight of clinical final formulation testing. Ensuring that clinical final formulation testing is both designed and conducted in a manner that will yield reliable results is critical, as is ensuring the protection of the human subjects on whom SPF and water resistance testing are conducted. Existing provisions within the SPF test in § 201.327(i)(3)(iv) require that informed consent be obtained, but do not otherwise specify what this should involve or how clinical final formulation testing should be overseen. Across disciplines, testing involving human subjects is ordinarily conducted under institutional review board (IRB) oversight as a means of ensuring that informed consent and other human subject protections are provided and ensuring the integrity of study design and execution. FDA likewise expects that IRB review is already routinely being obtained by many establishments for SPF and water resistance testing.
Nonetheless, our experience in conducting inspections and other actions to verify the reliability of final formulation testing under the current provisions of § 201.327 have raised some questions about current practices. For example, FDA's observations have raised questions about whether and how entities conducting final formulation testing have put in place protocols and IRB oversight to ensure that test subjects do not repeat participation in testing with a frequency that could both compromise the ability to distinguish erythemic reactions to the test article and raise other questions about human subject protection. We are concerned that the lack of explicit requirements with regard to IRB oversight, as well as the cursory nature of the informed consent requirement in the current sunscreen labeling regulation, may result in inconsistent practices in the conduct of SPF testing that would compromise the reliability of results. Among other things, IRB review is critical to verify the adequacy of informed consent and to ensure that study protocols incorporate appropriate inclusion/exclusion criteria for subject selection (both to protect test subjects and to ensure the accuracy of results).
b. Qualifications of study personnel. In some instances, it may not be clear upon inspection whether all aspects of a study were conducted by appropriately qualified personnel. For example, FDA would not consider it adequate for a technician, rather than an appropriately trained medical professional (such as, for example, a nurse or dermatologist), to perform a physical examination for potential nevi, moles, or other dermal lesions. As with all clinical and nonclinical testing done to support labeling, the use of properly trained and appropriately qualified personnel is essential to ensure the reliability and accuracy of test results. Documentation of the qualifications and training of personnel is also necessary to enable FDA's efficient enforcement of the FD&C Act.
c. Documentation of equipment maintenance, study methods, and observations. Failure to maintain adequate records of testing equipment, methods, and observations can raise broad questions about the reliability of final formulation testing. In FDA's experience since the promulgation of current § 201.327, there has been a lack of uniformity in testing entities' approaches to recordkeeping for final formulation testing, raising concerns about the adequacy of recordkeeping procedures. Failure of testing entities to keep adequate records to support final formulation testing may leave FDA unable to verify that the UV doses provided in SPF and water resistance test reports are accurate and valid. This is also true with respect to documentation of emission spectrum, the percentage of erythema-effective radiation contribution, and changes to solar simulator components and the UV meter/dose controller system. Failure to accurately calibrate and maintain equipment at one testing entity may affect data across multiple clinical SPF testing studies and/or broad spectrum testing for multiple different final formulations that are ultimately sold under different labels. Inadequate recordkeeping may interfere with efficient enforcement. We propose to address these concerns and align the regulation with our existing expectations through revised regulatory provisions that are described further in the following sections.
2. Specific Regulatory Proposals
a. Consequences of failure to observe best practices. We propose to clarify in the introductory paragraph of § 201.327 that a product is deemed misbranded if its labeling relies on the results of final formulation testing that was not conducted in compliance with all of the applicable provisions of § 201.327. Unless testing is conducted in compliance with all applicable provisions of § 201.327, FDA does not have adequate assurance that the labeling reliably reflects the properties of the sunscreen product. Therefore, if final formulation testing is not properly conducted in accordance with this section, labeling a sunscreen with an SPF value or representation of water resistance or broad spectrum properties based on that testing is a misrepresentation to the consumer that the labeling reliably states the product's properties, which should also be consistent with a system of standardized sunscreen labeling that can be used to make cross-product comparisons. We propose to incorporate the provisions of § 201.327(a) through (l) into part 352 as conditions under which a sunscreen is GRASE and not misbranded. If these provisions are finalized, failure to comply with these conditions would make a drug subject to regulatory action as misbranded and an unapproved new drug.
b. General obligations of responsible persons. We are aware that many different business relationships involving numerous entities are commonly used in the manufacturing, testing, and labeling of nonprescription sunscreen drug products. To clarify the locus of responsibility for ensuring that adequate final formulation testing procedures are in place, and to clearly delineate responsibility for Start Printed Page 6241recordkeeping related to final formulation testing, FDA proposes a new defined term, responsible person.
We propose to define the term responsible person in a way that is consistent with FDA's treatment of regulatory responsibilities for other OTC drug products and that is in alignment with requirements for adverse event reporting for over-the-counter drug products, in section 760(b)(1) of the FD&C Act. The proposed definition for responsible person is “the manufacturer, packer, or distributor whose name appears on the labeling of a sunscreen product covered by this section.” Defining responsible person in this way will enable FDA to better assess compliance with § 201.327 because it creates a chain of responsibility that is immediately apparent from the product's labeling. The responsible person, as identified on the labeling, is ultimately responsible for ensuring that the product bearing its name is labeled in accordance with the requirements of § 201.327.
The proposed revision of § 201.327(a) would broadly set forth the general obligations of responsible persons with respect to final formulation testing under § 201.327(i) and (j), and it would make clear that the responsible person is charged with ensuring that sunscreen products are appropriately tested. The obligations of responsible persons as enumerated in § 201.327 are modeled after those of investigational new drug application (IND) sponsors under part 312 (21 CFR part 312), but are somewhat modified to accommodate unique aspects of clinical and nonclinical sunscreen formulation testing. Because final formulation testing under § 201.327(i) and (j) is intended to verify the claimed properties of a final formulation, and because this purpose is narrower in scope and duration than most clinical testing performed under FDA's IND regulations in part 312, a responsible person under proposed § 201.327 would have responsibilities that incorporate some of the traditional responsibilities of investigators as well as those of sponsors under part 312. For example, FDA proposes to clarify that responsible persons must select appropriately qualified personnel to conduct testing, ensure compliance with the requirements for IRB review and obtaining informed consent, and monitor the compliance of personnel with investigators' statements.
This proposed approach accounts for situations in which investigators and other personnel conducting final formulation testing are employees of the responsible person. We also propose to clarify that the responsible person must ensure that investigators and other personnel conducting investigations under § 201.327(i) comply with requirements related to human subject protection and the appropriate conduct of clinical testing. We believe that this better reflects the employer/employee relationships that are more common in connection with final formulation testing rather than with clinical testing conducted under an IND. These proposed provisions regarding selection of personnel are also consistent with the existing obligations of manufacturers under parts 210 and 211 (21 CFR parts 210 and 211), both of which govern compliance with CGMPs.
The proposed revision of § 201.327(a)(1) permits a responsible person to transfer some or all of its obligations to another entity, consistent with current industry practice, except for obligations with respect to recordkeeping. The recordkeeping proposal is discussed in section IX.D. Failure of an entity to comply with provisions of this part governing responsibilities it has assumed would subject that entity to the same regulatory action as if it were a responsible person who had failed to comply with those obligations. This provision is analogous to the provision in FDA's regulations at part 312 allowing for transfer of obligations of IND sponsors.
c. Adequate clinical testing procedures and conditions. Although current § 201.327 requires “legally effective written informed consent from all test subjects” (§ 201.327(i)(3)(iv)), it does not address broader underlying requirements for conducting clinical testing. In light of the concerns we identified regarding current clinical testing procedures and conditions, we propose to amend § 201.327 by adding paragraph (i)(1), “Adequate Clinical Testing Procedures and Conditions.” We expect that final formulation testing conducted in compliance with the proposals in this paragraph will be more likely to ensure protection of human subjects while also more reliably determining the SPF value and water resistance properties of the final formulations being tested. Unless appropriate clinical testing procedures and conditions are adhered to, FDA cannot have confidence in the resulting labeled SPF and water resistance properties of the product.
Proposed § 201.327(i)1(B) and (C) have been added to make clear that FDA's regulations governing informed consent (part 50 (21 CFR part 50)) and IRB approval of research (part 56 (21 CFR part 56)) apply to clinical final formulation testing that is conducted under § 201.327(i). In our view, as a matter of good clinical practice, IRB approval should already be routinely currently obtained for clinical final formulation testing under current § 201.327 because it is essential to producing results that are scientifically sound and ethically appropriate. Because clinical final formulation testing required to support labeling under current § 201.327 is not conducted under an IND or in support of a GRASE determination in the OTC sunscreen monograph, it was not previously included explicitly in the scope of testing covered by parts 50 and 56. We propose to rectify this omission by explicitly cross-referencing parts 50 and 56 in revised § 201.327(i). This will clarify that both of these parts apply to clinical final formulation testing and will resolve any inconsistency in current practice.
The proposed reference to part 50 clarifies FDA's position that legally effective written informed consent to participate in clinical final formulation testing should share the same properties as informed consent required for all other clinical testing covered by FDA's regulations in part 50. Similarly, by referencing part 56, the proposal ensures that final formulation testing is held to the same standards for IRB review as other clinical testing covered by FDA's regulations. In reviewing clinical protocols, IRBs have the ability to determine whether the protocol is adequately designed to study the endpoints sought, and to ensure that protocol elements, such as enrollment criteria, adequately protect both human subjects and the scientific rigor of the experiment.
d. Control of personnel. We propose to place responsibility on the responsible person to ensure that investigators and other personnel conducting clinical final formulation testing adhere to the investigational plan, the signed investigator statement, and all applicable regulations. We also propose to place responsibility on the responsible person for ensuring human subjects' protection, including through appropriately reporting changes in the testing to IRBs, and by appropriately seeking prior IRB approval for any changes to the testing, except where necessary to eliminate apparent immediate hazards to human subjects. Under the proposed rule, responsible persons are also expected to obtain from each investigator, and retain for their records, a signed investigator statement. This is similar to what is required of sponsors of INDs, and it helps to ensure that the investigator is qualified, understands his or her obligations, and will comply with the requirements of Start Printed Page 6242this paragraph and with the protocol. It also enables better oversight of clinical investigations by FDA because it creates a record of the investigator's relevant experience and qualifications.
e. Research monitoring. A number of changes in § 201.327(i)(1) are being proposed to ensure adequate monitoring of clinical final formulation testing. Revised § 201.327(i)(1) would require that responsible parties inform all investigators testing a formulation if there are new observations about the drug, particularly with regard to adverse events or safe use. This is necessary to ensure proper communication between study personnel and protection of human subjects. Responsible persons must also monitor the conduct of investigations to ensure that clinical testing is being conducted in accordance with the protocol and with applicable regulations. If a responsible person discovers noncompliance by study personnel, then the responsible person must either secure compliance or remove the noncompliant personnel from conducting testing.
Finally, we propose to require that investigators report adverse events and/or safety concerns to the responsible person, and that investigators also provide responsible persons with final reports at the conclusion of testing. We believe that this will ensure there is appropriate documentation and communication of adverse events and/or safety concerns that arise during testing. It will also ensure there is a record of SPF testing conducted under § 201.327(i) that can be relied upon should questions related to a particular formulation arise when the sunscreen formulation is marketed. The proposed requirements are consistent with reporting required in the IND context, although, because of the short duration of the clinical final formulation testing conducted under § 201.327(i), we are not proposing to require annual reporting.
f. Test subject selection. We propose additional language regarding the selection of test subjects in § 201.327(i)(4). This is an area in which FDA's inspections of testing entities have suggested a lack of consistency. We are particularly concerned that inclusion/exclusion criteria provide for adequate time between study and enrollment and prior UV exposure, such as from participation in a previous SPF test, sunbathing, or sunlamp use. Erythemal responses can remain for days after sunbathing, and it is known that pigmentation development takes up to a week after initial exposure and remains for weeks to months (Ref. 186). SPF clinical studies should not include individuals who have participated in sunbathing, tanning bed use, or another SPF clinical study for at least the past 4 weeks or perhaps longer if UV-induced responses remain. The proposed clarification regarding conduct of physical examinations of test subjects reflects this consideration, and our additional proposal for IRB review, addressed elsewhere, will help ensure it is appropriately acted on.
g. Applicability of registration and CGMP requirements. Proposed § 201.327(k) reflects FDA's existing view that final formulation testing conducted under § 201.327 constitutes the “manufacture” of a drug. As such, this testing must be conducted in an establishment registered in accordance with part 207 (21 CFR part 207) and section 510 of the FD&C Act. This interpretation is consistent with the definition of manufacture in part 207, which includes “each step in the manufacture, preparation, propagation, compounding, or processing of a drug . . . .” (§ 207.1). The definition of manufacture as used in part 207 also “includes manipulation, sampling, testing, or control procedures applied to the final product or to any part of the process, including, for example, analytical testing of drugs for another registered establishment's drug” (id). Accordingly, a sunscreen product labeled in reliance on final formulation testing done in an unregistered establishment is misbranded under section 502(o) of the FD&C Act. This interpretation is also consistent with FDA's regulations in § 330.1, which require that OTC monograph drug products be manufactured in a registered establishment in order to be generally recognized as safe and effective and not misbranded. The incorporation of this provision in § 201.327, therefore, is intended to clarify an existing requirement for facilities performing this type of testing.
We also propose to clarify that, as a manufacturing activity, final formulation testing conducted under this paragraph is expected to be done in accordance with CGMPs as set forth in parts 210 and 211 (see § 210.3(b)(12), indicating that for the purposes of parts 210 and 211, “Manufacture, processing, packing or holding of a drug product includes packaging and labeling operations, testing, and quality control of drug products”). This is consistent with FDA's regulations in § 330.1, which require compliance with CGMPs as a condition for OTC drug products to be GRASE and not misbranded when otherwise marketed consistent with conditions in a final monograph. Adherence to CGMP requirements in parts 210 and 211 includes compliance with the requirements to keep certain records and to have appropriately trained and qualified personnel. Failure to comply with CGMPs results in a product being adulterated under section 501(a)(2)(B) of the FD&C Act.
h. Recordkeeping. To enable FDA to better monitor compliance with the requirements of § 201.327, we propose to include specific recordkeeping requirements for final formulation testing. Accordingly, proposed § 201.327(l) clarifies what records of testing performed under this section must be kept, by whom, and for how long. This provision also allocates responsibility for records maintenance and specifies what records must be made available to FDA for inspection. Recordkeeping is essential for FDA to evaluate whether required testing of final formulations is being conducted in accordance with § 201.327(i) and (j), and to enable the Agency to investigate postmarketing product failures or adverse events. Appropriate recordkeeping also enables FDA to conduct better and more efficient inspections of entities conducting final formulation testing.
These recordkeeping requirements are in alignment with what is required for other types of manufacturing under CGMPs as set forth in parts 210 and 211. The proposed provisions are intended to clarify how, and for how long, records must be kept to substantiate required final formulation testing. We are proposing that records of testing must be kept by the responsible person (as newly defined in § 201.327(a), discussed previously), as well as by any other entity that actually performs testing (under a transfer of obligations per § 201.327(a)(1) or otherwise). Requiring that records be kept by both the responsible person and the testing entity (if different) will enable FDA to more easily identify records supporting the labeling of any given final formulation even when the product is labeled with the responsible person's information, but testing and manufacturing was completed by a third party.
The proposed recordkeeping requirements reflect FDA's experience in interacting with regulated industry. By requiring that records be kept by both the responsible person and any other entity that performs final formulation testing, the proposed rule will enable more efficient enforcement of the FD&C Act by, for example, allowing FDA to identify the source of formulation failures or apparent inconsistencies between the product labeling and consumer experience. The proposed recordkeeping requirements Start Printed Page 6243will also assist FDA when it is conducting inspections of entities that perform final formulation testing for a number of different responsible persons and products, as we believe is the norm in this industry. Having ready access to records reflecting the overall conduct of final formulation testing during an inspection of such an entity is important because it will enable FDA to identify potential systemic problems in final formulation testing that may have an impact on the reliability of results supporting the labeling of multiple different sunscreen products marketed by a variety of responsible persons. We note that these recordkeeping requirements should not be understood to mandate duplicative records within the files of a single testing entity or single responsible party. For example, if one investigator is responsible for testing multiple final formulations, one copy of the signed investigator statement and Curriculum Vitae (CV) would be sufficient to support all formulations tested by that investigator.
Consistent with FDA's view that final formulation testing is manufacturing, and thus is subject to CGMPs, equipment maintenance records and other records documenting compliance with CGMPs are expected to be maintained as required by parts 210 and 211. Accordingly, we clarify in proposed § 201.327(l) that records documenting proper maintenance of equipment used in final formulation testing must be kept, consistent with existing obligations in 21 CFR 211.68. In our view, this clarification will promote uniformity in adherence to best practices and will help ensure more accurate and reliable labeling of sunscreen products based on final formulation testing. Additional specificity has been proposed here to clarify how the more general recordkeeping provisions of part 211 apply to final formulation testing. To provide assurance that the test results are not compromised by faulty equipment maintenance or equipment failure, FDA proposes that testing entities must keep documentation demonstrating that equipment used for final formulation testing has been maintained in accordance with established written specifications. This requirement will enable FDA to more efficiently monitor compliance. Failure to keep required records of final formulation testing will render a product whose labeling relies on that testing adulterated under section 501(a)(2)(B) of the FD&C Act. Without recordkeeping, there is no assurance that a sunscreen drug product has the identity and strength, and meets the quality and purity characteristics, which it purports or is represented to possess.
This proposal also elaborates on recordkeeping necessary to document compliance with the requirements of proposed § 201.327 regarding conduct of final formulation testing.
Proposed required records for SPF testing include records that: (1) Identify the facility conducting the testing; (2) identify the equipment used; (3) identify product samples and lots; (4) characterize the SPF standard that is used; (5) document parameters for water resistance testing; and (6) demonstrate compliance with the provisions governing adequate clinical testing procedures and conditions. For example, these would include documentation of IRB review, case histories for each human subject (which must document protocol deviations or injuries), administration of the sunscreen, and reading of test results. These proposed recordkeeping obligations are consistent with those required of parties engaged in human subjects testing governed by other portions of FDA's regulations.
Required records of broad spectrum testing conducted under proposed § 201.327(j) would include those records necessary for identifying the facility conducting the testing, providing information associated with the sample, identifying equipment used, and documenting sunscreen product application. These proposed requirements provide greater specificity than existing requirements in FDA's CGMP regulations, and are expected to increase uniformity in current practice.
We propose to clarify FDA's expectations regarding access to records that responsible persons and other testing entities are required to keep under this paragraph. These provisions are consistent with FDA's inspection authorities in section 704 of the FD&C Act.
i. Minor proposed revisions to test procedures. In addition to the changes discussed in section IX.D, we are proposing several modifications to the technical instructions for sunscreen final formulation testing (§ 201.327(i) and (j)) to clarify how the testing should be conducted.
We are concerned that manufacturers conducting the SPF test procedure may be relying on determinations of the initial minimal erythema dose of unprotected skin (MEDu) generated too far in advance of testing the sunscreen product. The current regulation in § 201.327(i)(5) addresses four different determinations of MED for each test subject: (1) An initial MED for unprotected skin (initial MEDu); (2) a final MED for unprotected skin (final MEDu); (3) an MED for skin to which the SPF standard has been applied (ssMEDp); and (4) an MED for skin to which the sunscreen test product has been applied (tpMEDp). The initial MEDu is used to set the UV exposures administered to determine final MEDu, ssMEDp, and tpMEDp (see § 201.327(i)(5)(iii)).
Although the regulation already requires that each of the MED values be determined 16 to 24 hours after UV exposure, it merely notes that the final MEDu, ssMEDp, and tpMEDp are “typically determined the day following determination of the initial MEDu” (see current § 201.327(i)(5)(iv)). Because the skin reactivity of a test subject changes over time, we propose to clarify that the initial MEDu of a person's unprotected skin must be determined no more than 1 day before the UV exposures for final MEDu, ssMEDp, and tpMEDp are administered. We are also clarifying that to calculate the SPF value for each test subject, under proposed paragraph § 201.327(i)(6), it is the subject's final MEDu that should be used.
In our review of the testing requirements as part of this rulemaking, we also revisited our position on the input slit bandwidth specification in the in vitro broad spectrum test. In the 2011 L&E Final Rule, we modified the in vitro broad spectrum test that was proposed in the 2007 proposed rule to change the input slit spectrometer bandwidth specification from ≤5 nm to ≤1 nm. After the 2011 final rule published, FDA received a comment from a spectrometer manufacturer arguing that the 1 nm input slit bandwidth specification was unreasonable. The manufacturer argued that common spectrometer models that are currently used to test sunscreens cannot comply with the ≤1 nm input slit bandwidth specification, and those that can are more expensive, more difficult to use, and take more time to use. The manufacturer provided data that indicate that spectrometers with ≤1 nm input slit bandwidths do not produce more reliable results than spectrometers with larger input slit bandwidths (see Comment, Docket No. FDA-2010-D-0509-0004). In light of this submission, FDA reassessed the input slit bandwidth parameters and concluded that ≤5 nm will be sufficient for the broad spectrum procedure. Although decreasing bandwidth improves the ability to resolve closely spaced peaks (i.e., the spectral resolution), this is not a significant consideration for in vitro broad spectrum testing of sunscreen products because transmittance/absorbance curves for sunscreen Start Printed Page 6244products are typically smooth with no individual sharp peaks. Accordingly, we propose to revise § 201.327(j)(1)(iv) to require that spectrometer input slits be set to provide a bandwidth that is ≤5 nm.
Establishing standardized testing procedures for sunscreen products and basing the products' labeling on this testing not only helps assure the safety and effectiveness of each product, it also provides consumers with consistent information about the sun protection properties of sunscreen products across brands, which in turn facilitates consumer comparisons when selecting products. Accordingly, we propose to delete the provision in § 352.77 (21 CFR 352.77) addressing test modifications or alternative testing procedures. Section 352.77 indicates that such test modifications or alternative testing procedures require submission of a petition in accordance with § 10.30 (21 CFR 10.30). The proposed removal of § 352.77 does not alter the existing ability of a firm or individual to petition the Agency to amend the monograph (see §§ 330.10(a)(12) and 10.30) to change the conditions that apply to products marketed under its provisions, such as to modify testing procedures for all products having some particular set of characteristics. Rather, the proposed deletion will clarify that the sunscreen monograph does not permit variation for individual products from the standardized testing procedures that are monograph conditions, because such variation could undermine important values supported by standardization.
We are also proposing to correct a minor inaccuracy in the existing regulatory language describing testing procedures. Specifically, § 201.327(i)(1)(ii)(C) currently states that “emission spectrum must be determined using a handheld radiometer.” As written, this statement is inaccurate because a handheld radiometer cannot determine the emission spectrum of a solar simulator. We propose to resolve this error by clarifying that the handheld radiometer measures the solar simulator radiation intensity rather than the emission spectrum. Finally, we have proposed edits to certain provisions describing final formulation testing procedures to clarify our long-standing intention that these provisions of the test are requirements, not merely suggestions.
E. Proposed Status of Sunscreen-Insect Repellent Combination Products
1. Background
Sunscreen-insect repellent combination drugs are products used on human skin that contain both a sunscreen drug component and an insect repellent component. A list of insect repellent products on the EPA website identified a number of such products as of November 2017 (including multiple products within a single brand line).[47] Among those products, the majority contained either N,N-Diethyl-meta-toluamide (also called DEET) or IR3535 as the insect repellant, and a few contained oil of citronella as the insect repellent. Combination insect repellent-sunscreen products have been marketed in a variety of dosage forms (see section IX.A for a discussion of dosage forms), with labeled SPF levels ranging from 15 to 30 (Ref. 187). Some products are also labeled as water resistant or very water resistant (Ref. 187). The products are generally labeled for use without regard to age (Ref. 187).
FDA regulates sunscreens as drug products under the FD&C Act, and EPA concurrently regulates insect repellents as pesticides under FIFRA.[48] FIFRA defines a “pesticide” in relevant part as “any substance . . . intended for repelling . . . any pest,” including insects (7 U.S.C. 136)(u)). Before they can be marketed, most skin-applied insect repellents must be registered by EPA, although a few plant-derived insect repellent active ingredients are exempt from registration because EPA has determined they present minimum risk potential to humans (Ref. 189).
Sunscreen-insect repellent combination products have been marketed in the United States since before the OTC review began, but they have not previously been addressed in the rulemaking for the OTC sunscreen monograph (72 FR 7941 at 7943). Both FDA and EPA have historically declined to object to the marketing of these products pending the issuance of a final sunscreen monograph, provided that the sunscreen active ingredient(s) is listed in the stayed final monograph and the insect repellent component is registered with the EPA (79 FR 7941 at 7943). In 2011, FDA issued a draft enforcement guidance intended for manufacturers who market OTC sunscreen products without an approved application, which recommended that manufacturers of sunscreen-insect repellent combination products should comply as closely as possible with FDA's sunscreen testing and labeling requirements in § 201.327. This guidance was finalized in May 2018 (Ref. 1).
In the Federal Register of February 22, 2007 (72 FR 7941), FDA issued a notice seeking public comments on sunscreen-insect repellent combination products, and, in particular, whether FDA should amend the OTC sunscreen monograph to add conditions for marketing insect repellent-sunscreen drug products (FDA Call for Data or call for data). The call for data summarized the regulatory status and history of both sunscreens and insect repellents, and sought public input on a number of issues (see table 6). On that same date (February 22, 2007, 79 FR 7979), EPA published a similar notice announcing that it was also seeking information to determine how insect repellent-sunscreen combination products should be regulated.
Table 6—Key Issues and Information Requests in FDA's 2007 Call for Data
General issue Key concerns and information requests Possible manufacturing conflicts Requested information about whether there are known conflicts between FDA and EPA manufacturing requirements and, if so, how to resolve them. Asked how FDA should address EPA-registered insect repellents in finalizing the OTC sunscreen monograph; which requirements should FDA retain, revise, or eliminate? Inquired about manufacturer testing of sunscreen-insect repellent combination products and whether any problems were encountered. Possible formulation conflicts Requested comments on the significance of published research suggesting a potential formulation conflict. Start Printed Page 6245 Possible labeling conflicts between OTC sunscreen monograph and EPA registration requirements Note: Since publication of the call for data, FDA has established additional labeling regulations for certain OTC sunscreen products marketed without approved applications. However, the labeling concerns expressed in the call for data remain relevant. Labeling differences noted: • FDA uses “warning”; EPA uses “caution” (and only uses the word “warning” to indicate toxicity levels). • Many differences in required warning/caution section headings. • Directions for sunscreen use call for liberal application and frequent reapplication; EPA directions may limit where and how to apply product and restrict frequency of application. Asked whether different directions for use can be integrated without leading to improper application, overexposure to insect repellent, and/or underexposure to sunscreen. FDA requires the outside container or wrapper of the retail package or the immediate container label to list all active and inactive ingredients (see section 502(e)(1)(A)(iii) of the FD&C Act; § 201.66(c)). EPA requires listing of the percentage of each active ingredient, and the total percentage of all “inert” or “other” ingredients, in the pesticide. Inert ingredients are not required to be identified individually on the product except in certain cases (in which case all inert ingredients are listed). Asked whether there is a way to label combination sunscreen-insect repellent drug products in a way that satisfies both the requirements of the FD&C Act and the FIFRA, and whether “inert” ingredients under the FIFRA are equivalent to “inactive” ingredients under the FD&C Act. Safety issues More safety data needed given published animal studies indicating increased absorption of DEET and various sunscreens active ingredients when the components are combined. Asked for more safety data on combined products. Requested data on whether increased absorption of a sunscreen ingredient occurs when combined with an insect repellent. Information needed about incidence of skin irritation from combination products. Effectiveness issues Requested information on: • Possible effects of insect repellent on sunscreen SPF; possible decreased sunscreen efficacy or increased exposure to insect repellent without greater efficacy resulting from inconsistent reapplication intervals. • Potential chemical or physical incompatibilities between particular sunscreens and insect repellents. • Potential need to specify minimum SPF for these combinations. • Any potential performance benefits of these combination products other than convenience. • Possible adjustments to formulations to minimize application time disparities. 2. FDA's Evaluation of Sunscreen-Insect Repellent Combination Products
FDA has reviewed the comments submitted in response to FDA's and EPA's calls for data, as well as pertinent scientific literature and publicly available EPA regulatory documents. Based on that review, we have tentatively concluded that sunscreen-insect repellent combination products, as a class, are not GRASE (i.e., are Category II) and are misbranded because conflicting labeling requirements for their sunscreen and insect repellent components cannot be reconciled to create labeling that will sufficiently ensure safe and effective use of the sunscreen component, as well as adequate directions for use as a sunscreen, as required by section 502(f) of the FD&C Act. Also, if we did not have this labeling concern, we would still tentatively determine that available data regarding the safety and effectiveness of these products for their use as sunscreens are insufficient to classify these sunscreen products as GRASE for such use (i.e., Category III). Specifically, evidence suggests that interactions between some sunscreen active ingredients and insect repellents may decrease safety by increasing systemic absorption of one or both components, and potential synergistic effects on the efficacy of sunscreen active ingredients apparently have not been studied. Although the available data are limited and not conclusive, they give rise to questions about the safety and effectiveness of these products. Our reasons for these tentative conclusions are detailed in the discussion that follows.
a. Public comments on the 2007 call for data. FDA received six submissions in response to the 2007 call for data. None of the comments included substantive data, although some cited published scientific and medical literature, which is addressed in the following section of this document. Five of the six comments were from manufacturers or a trade association. Industry comments generally favored retaining joint regulation between EPA and FDA (perhaps with enhanced coordination and information-sharing) and amending the stayed OTC sunscreen monograph to address sunscreen-insect repellent combinations. Several industry comments claimed there was an absence of conflicting requirements relating to manufacturing, formulation, and/or labeling. Others suggested approaches for minimizing labeling conflicts, such as permitting exemptions to FDA's Drug Facts labeling requirements to accommodate EPA-required information, or placing FDA- and EPA-required information in separate areas of the label. The remaining comment was submitted by a medical association that opposed continued marketing of sunscreen-insect repellent products, emphasizing concerns about children's exposure to DEET. Industry comments favoring the continued marketing of combination sunscreen-insect repellent drug products also contended that combining sunscreen and insect repellent ingredients in a single product is more convenient and cost-effective than using separate products. Two comments stated that properly formulated, tested, and labeled, combination products are better than the unpredictable effects that could arise when consumers use two different products. Regarding safety, one comment asserted various flaws in the studies cited in the call for data that questioned the safety of these combination products. (These studies are discussed in section IX.E.2.d.)
In general, the comments that we received in response to the 2007 call for data were not accompanied or corroborated by data. Although the comments did not identify further concerns relating to product manufacturing or formulation, they did not adequately address FDA's concerns about safety, effectiveness, and labeling of these products. FDA renews its request for data to support labeling and safety for sunscreens with insect repellent added.
b. Pesticide-related information. Pesticides that are or have been used in combination products that also contain sunscreens include DEET, IR3535, and oil of citronella. In evaluating combination insect repellent-sunscreen products for the purposes of this rule, FDA defers to EPA's expertise and authority regarding insect repellent ingredients. We have not independently evaluated these pesticides, but instead have focused on potential sunscreen-insect repellent ingredient interactions and the feasibility of effectively labeling these combination products for their use as sunscreens.Start Printed Page 6246
As of June 2017, DEET was by far the most commonly used insect repellent. According to the EPA Product list, the amount of DEET in combination sunscreen-insect repellent products ranged from 10 to 20 percent. DEET product labels recommend that users avoid over-application, use just enough repellent to cover exposed skin and/or clothing, and not apply to hands or near the eyes or mouth of young children (Ref. 190). DEET-containing products listed on the EPA website in 2017 had concentrations ranging from 5 percent to 98 percent and provided protection from mosquitos for 2 to 12 hours, with many products having protection times of 4 hours or more (Ref. 187). The American Academy of Pediatrics recommends that repellents should contain no more than 30 percent DEET when used on children, and that insect repellent should not be used on children younger than 2 months (Ref. 191).
EPA classifies the acute toxicity of insect repellents and other pesticides into one of four toxicity categories (ranging from Category I, highly toxic, to Category IV, practically nontoxic) (see 40 FR 156.62). DEET is classified in Category III based on EPA's review of available animal studies, indicating slight acute toxicity for acute oral, dermal, ocular, and inhalation tests in animals, and low acute toxicity for the human health risk assessment (Ref. 192). Although DEET is registered for use in humans of any age, adverse events related to DEET toxicity have been documented and these events primarily relate to the central nervous system. As summarized by Katz et al., DEET has been associated with seizures and other central nervous system symptoms, cardiovascular symptoms, and topical and allergic symptoms (Ref. 193). Most reported cases of adverse or lethal events involved overuse or otherwise incorrect use of the product (Ref. 193), and EPA concluded that available data were insufficient to identify DEET as the cause of the reported adverse events (Ref. 192). EPA is currently in the process of updating its registration of a number of older pesticides, including DEET, and is deferring decision on the regulatory status of combination DEET/sunscreen products as described in the EPA Call for Data.[49] However, EPA has stated that DEET does not pose a significant health risk to the U.S. population.[50]
IR3535 is classified by EPA as a biopesticide because it is biochemically, functionally identical to beta-alanine, a naturally occurring substance that repels insects (Ref. 194). IR3535 is classified in Toxicity Category IV (practically nontoxic) for acute oral, dermal, and inhalation toxicity and Category III (slightly toxic) for eye irritation (Ref. 195). Overall, EPA has assessed IR3535 as not harmful when ingested, inhaled, or used on skin (Ref. 195). Eye irritation could occur if the chemical enters a person's eyes (Ref. 195). IR3535 is used at concentrations of 7.5 percent to 20 percent in a popular line of sunscreen-insect repellent combination products (EPA Product List) (Ref. 187). Products containing IR3535 identified on EPA's website in summer 2017 had concentrations ranging from 7.5 percent to approximately 20 percent and listed protection time against mosquitoes of 2 to 8 hours (EPA Product List) (Ref. 187).
Oil of citronella is a plant-derived biochemical insect repellent (72 FR 7979 at 7981). Depending on its source, it may be categorized as “Ceylon” type or “Java” type. It is currently listed by EPA as a minimum risk pesticide (registration generally not required if formulated only with EPA-permitted inert ingredients and not labeled as effective against disease-causing pests) (40 FR 152.25(f)). Oil of citronella is also an approved food additive for use as a flavoring agent in foods and beverages (Ref. 196). EPA has designated oil of citronella as Toxicity Category III (slightly toxic) for acute oral toxicity (Java type only), dermal toxicity, dermal irritation, and acute eye irritation (both types), and Category IV (practically nontoxic) for acute oral toxicity (Ceylon type) and acute inhalation (Ref. 197). The National Pesticide Information Center (NPIC) fact sheet on oil of citronella states that oil of citronella products should not be used on children less than 6 months old (Ref. 198).
c. Disparities in required labeling of sunscreens and insect repellents. FDA and EPA regulate the format and content of the labeling of nonprescription sunscreen products and pesticides, respectively. FDA regulations on nonprescription sunscreen labeling include the general drug labeling regulations in subpart A of part 201; the “Drug Facts” format and other OTC drug labeling requirements in subpart B of part 201; and the sunscreen-specific labeling requirements that apply to sunscreens marketed without an approved NDA, including those based on the current requirements for SPF and broad spectrum testing in § 201.327. The labeling of registered insect repellents is subject to EPA labeling requirements under FIFRA (40 CFR 156), as well as specific language specified in individual product registration documents. Although the FDA and EPA labeling requirements for nonprescription sunscreens and registered pesticides cover some of the same information (such as ingredient lists, net quantity statements, and warnings/precautions), there is considerable variation in the language, format, and placement of common label elements between the two Agencies, while other elements do not overlap.
Furthermore, both Agencies limit the degree to which a drug manufacturer or pesticide registrant may depart from the prescribed text, format, and/or location of required labeling elements. This is particularly true for the wording and format of “drug facts” information for OTC drugs (see § 201.66). Similarly, EPA regulations state that although a registrant may choose to place non-FIFRA-required information on a pesticide label, it may not replace, obscure, conflict with, or supersede the FIFRA-required text (Ref. 199).
The intended uses of sunscreens and insect repellents are quite different, as are the associated labeling requirements; in particular, the instructions for using the two types of products are different. Required labeling for OTC sunscreens marketed without approved NDAs calls for reapplication at least every 2 hours (see § 201.327(e)(3) through (e)(4)). The duration of protection for insect repellents varies according to the active ingredient and strength. Based on information from the EPA product list, many insect repellent-sunscreen products provide protection against mosquitoes and/or ticks for more than 2 hours, and some provide protection for as many as 6 to 10 hours. EPA has stated that it is “concerned about consumer use of products that contain sunscreens and DEET, since directions to reapply generally and frequently may promote greater use of DEET than needed for pesticidal efficacy and thus pose unnecessary exposure to DEET.[51] The Centers for Disease Control and Prevention (CDC) advises consumers Start Printed Page 6247that “products that combine sunscreen and repellent are not recommended, because sunscreen may need to be reapplied more often and in larger amounts than needed for the repellent component to provide protection from biting insects.” (Ref. 200). Similarly, the American Academy of Pediatrics advises consumers not to use products that combine DEET with sunscreen, in part because “[t]hese products can overexpose your child to DEET because the sunscreen needs to be reapplied often” (Ref. 201). Additionally, DEET is approved for use on children with no age restriction (Ref. 202), whereas FDA labeling states “[bullet] children under 6 months of age, ask a doctor” (see § 201.327(e)(1)(iv)).
The recommended manner of application also differs for sunscreens and insect repellents. For example, the directions on the label for all insect repellent products containing DEET say to apply just enough to cover exposed skin, and avoid over-application (Ref. 190), whereas the labeling of nonprescription sunscreens marketed without approved NDAs calls for liberal or generous application (see § 201.327(e)(1)(ii)). The EPA-mandated directions on the labels of DEET products also state, “Do not apply near eyes and mouth; apply sparingly around ears; do not use under clothing” (Ref. 190). Such statements are potentially troublesome from the standpoint of sun protection in light of surveillance data from Australia, which suggest that the incidence of certain skin cancers is more frequent on highly exposed areas of the body such as ears and the backs of hands (Refs. 203 and 204). The CDC advises consumers who need protection from both sun and insects to apply sunscreen product first, followed by an insect repellent (Ref. 200).
Additional disparities in the content and format of labeling elements for sunscreens and registered insect repellents include the following:
- EPA pesticide labeling includes required elements that generally must appear on the front panel of the label, such as the ingredient statement (40 CFR 156.10(g)(2)), specified signal word such as “CAUTION” (40 CFR 156.64), and child hazard warning (40 CFR 156.66), which could crowd or detract from drug information required to appear on the principal display panel for drugs (see § 201.60 (“The principal display panel shall be large enough to accommodate all the mandatory label information required to be placed thereon by this part.”)). Other labeling elements that only EPA requires include registration numbers and manufacturing establishment numbers (40 CFR 156.10(a)).
- FDA labeling for sunscreens uses the word “warning” (see §§ 201.66(c)(5) and 201.327(d)), while the EPA requirements specify that pesticide products that, like DEET, IR3535, and oil of citronella, meet the criteria of Toxicity Category III or IV as the highest category by any route of exposure bear on the front panel either no signal word or only the signal word “CAUTION” (40 CFR 156.64). In EPA labeling the word “WARNING” is used as a signal word only for toxicity category II, which is a higher toxicity category than that applicable to any insect repellent ingredients used in sunscreen-insect repellent combination products (40 CFR 156.64(a)(2)).
- FDA labeling uses the term “directions” (see §§ 201.66(c)(6) and 201.327(e)), while EPA regulations use the term “directions for use” (see 40 CFR 156.10(i)(2)).
- FDA calls for ingredients to be listed as “active” and “inactive” (see § 201.66(b) through (c)), while EPA labeling uses the term “inert” or “other” instead of “inactive” for all non-pesticide ingredients (40 CFR 156.10(g)).
Given the extent of the disparities discussed above, FDA tentatively concludes that attempting to merge the required labeling for monograph sunscreens and insect repellents in a way that would comply with both Agencies' requirements and permit adequate consumer understanding and proper use would be impracticable. In this regard, we specifically disagree with comments made in response to the 2007 FDA Call for Data suggesting that acceptable “merged” labeling could be crafted by varying the OTC sunscreen drug facts to include insect-repellent-related information, and/or by providing EPA-required labeling outside the drug facts box. We are particularly concerned that consumers would be confused by the juxtaposition of two sets of different and, in some cases, contradictory information in the labeling about these products' dual intended uses. We are also concerned that the sheer amount of required information would result in crowded, difficult-to-read labels lacking in the clarity and prominence of important safety and use information that are both required by FDA regulations and vital to consumer comprehension. We solicit comment and data about how to reconcile the labeling of suncreens and insect repellents such that a combined product could meet FD&C Act requirements for OTC sunscreen drugs.
d. FDA's review of published medical literature. The results of FDA's literature review raise potential safety concerns about products that combine sunscreen and insect repellent active ingredients. The available data suggest that the dermal penetration and systemic absorption of at least one combination of a sunscreen active ingredient and an insect repellent is increased when both are present.
There have been some studies assessing the penetration of DEET and the effects of DEET combined with sunscreen (particularly the active ingredient oxybenzone) on dermal penetration. Ross et al. tested for synergistic effects between DEET and oxybenzone using an in vitro mouse skin diffusion model and showed substantial penetration of a 20 percent DEET standard in ethanol, while penetration of sunscreen active ingredients was not found (Ref. 205). Despite a lower DEET content (10 percent), a commercially marketed sunscreen formulation had a 6-fold more rapid detection and a 3- to 4-fold greater penetration of DEET than the 20 percent standard. Other diffusion tests using pigskin or artificial membranes and various combinations of DEET and oxybenzone in different media suggested an enhancing effect on dermal penetration of both DEET and oxybenzone (Refs. 206 and 207). The same investigators obtained similar results in a later in vitro study using human skin (Ref. 207).
Kasichayanula et al. assessed the dermal absorption of DEET and oxybenzone using an in vivo piglet model, in which samples were collected from plasma, urine, and under the skin. Their results indicated that the enhanced dermal penetration evidenced in the in vitro studies translated to increased systemic exposure to both oxybenzone and DEET (Refs. 208 and 209). Finally, a study by Yiin et al. suggests that enhanced systemic absorption would also occur in humans (Ref. 210). Yiin et al. used human urinary metabolites of DEET and oxybenzone to evaluate the mutual enhancing effect on absorption of these ingredients and concluded that their findings confirm that concurrent use of DEET-containing insect repellent and oxybenzone-containing sunscreen results in the enhancement of dermal absorption of DEET when insect repellent (DEET) was applied first and then covered by sunscreen (Ref. 210). The study authors suggested that placing repellent spray on top of sunscreen lotion with no mixing seems to be the best approach to diminish DEET penetration through the skin.
Although insect repellents and sunscreens are designed to exert their Start Printed Page 6248protective effects on the surface of the skin, the studies described above suggest that combining a sunscreen and insect repellent in a single product may result in unintended systemic exposure to the sunscreen ingredient oxybenzone and the insect repellent ingredient DEET. We acknowledge the study limitations cited by comments to the FDA Call for Data, and that in vitro diffusion studies have their limitations in terms of reflecting clinical use. We also note that many of the studies tested formulated commercial products with multiple sunscreen ingredients and excipients for which details were not given, and it is unclear how this may have influenced the results. Although we, therefore, do not view these data as conclusory, we have determined that they raise a valid safety concern that warrants a tentative conclusion that, even if one could overcome the misbranding and associated safety and effectiveness concerns created by the inconsistent application directions for sunscreens and insect repellants, there would not be sufficient evidence to conclude that combination sunscreen and insect repellent products are GRASE for sunscreen use without further investigation.
Regarding future investigations that could assist FDA in determining whether these products have sufficient evidence of safety to be GRASE for use as sunscreen, we are not aware of any data that define the extent of systemic exposure to either DEET or oxybenzone that would occur with maximal exposure to a sunscreen-insect repellent combination product. There also are few data from which to assess whether there would be a similar enhancement of skin penetration for other combinations of sunscreen and insect repellent active ingredients. Moreover, without adequate human absorption studies under maximal use conditions of particular sunscreen-insect repellent combinations (i.e., a MUsT, as discussed in section VII.B.4), it is difficult to evaluate potential risks associated with the use of such combination products. Because of the potential synergistic interaction between the sunscreen active ingredient and the insect repellent active ingredient, human absorption data for the individual components would not provide adequate data to estimate the level of systemic absorption. Likewise, in vitro data would not be able to provide a reliable estimate of the systemic exposure that would occur with such products' use.
In terms of sunscreen active ingredient effectiveness, we have little data from which to determine whether the presence of an insect repellent would affect the determined SPF value of combination sunscreen-insect repellent products. Montemarano et al. reported a reduction in sunscreen efficacy because of concomitant use with insect repellent. However, in that study, the sunscreen and insect repellent ingredients were applied separately and were not part of a combination product (Ref. 211).
With respect to efficacy, we recognize that the testing required by § 201.327 (both the current regulation and the regulation if amended as proposed elsewhere in this proposed rule) to support labeled SPF levels and other efficacy claims that may be made for certain OTC sunscreen products could potentially mitigate concerns about the impact of insect repellent active ingredients on sunscreen effectiveness. However, we are not aware of any data evaluating the reliability of SPF testing for sunscreen formulations that contain insect repellent ingredients. There also is the possibility that increasing the amount of the sunscreen active ingredient to compensate for any loss in efficacy because of the presence of the insect repellent could result in unnecessarily high exposure to the sunscreen active ingredient. For these additional reasons, we tentatively conclude that even if other concerns could be overcome, there is not currently sufficient evidence to conclude that combination sunscreen-insect repellent products are GRASE for use as sunscreens. We solicit comment on the data needs identified above and tentative conclusions, including supporting data and analysis. We also solicit data and information to address these data needs.
3. Conclusion
FDA tentatively concludes that the inherent disparity in labeling requirements that apply to sunscreens marketed under the OTC monograph and insect repellents prevent the creation of labeling that will sufficiently ensure safe and effective use of the sunscreen component of sunscreen-insect repellent combination products, particularly in connection with duration of action. We also conclude that these conflicting requirements prevent these products from having adequate directions for use as a sunscreen, and thus these products would be misbranded under section 502(f) of the FD&C Act. In addition, even if these issues could be overcome, existing safety concerns about potential enhanced systemic absorption resulting from combining individual sunscreen active ingredients and insect repellent ingredients would also need to be addressed by further studies on both combinations of individual sunscreen and insect repellent ingredients and final formulations.
Existing data indicates there is a risk of systemic absorption of insect repellent and/or a sunscreen active ingredient when both are present. Additional data would be needed to identify any interactions between specific sunscreen active ingredients and insect repellents, in particular, to characterize any enhancement of skin penetration and/or systemic absorption if the resulting data presents safety or effectiveness concerns. As stated above, FDA would need adequate human absorption studies, such as a MUsT, as part of the clinical safety assessment (for more discussion on assessment of dermal absorption of sunscreen active ingredients using MUsT, see section VII.B.4). The effectiveness of sunscreen-insect repellent combination products is also a continuing concern. For all of those reasons, we tentatively determine that these products are not GRASE for nonprescription sunscreen use. We solicit comment on this tentative determination.
X. Proposed Actions To Effectuate Lifting of Stay and Harmonize Impacted Regulations
In the 2011 L&E Final Rule, FDA explained that although we were not yet lifting the stay on the 1999 final monograph, the provisions set forth in the L&E Final Rule reflected the Agency's position on the appropriate testing and labeling of sunscreen products that were previously identified as falling within the Stayed 1999 Final Monograph (76 FR 35620 at 35621). We explained that § 201.327 would therefore supersede the prior approach to labeling and effectiveness testing described in the never-effective provisions of part 352, subparts C and D.
We are now proposing to lift the stay on the 1999 final monograph (21 CFR part 352) while making certain changes in its provisions. To fully effectuate this proposal, we are proposing several harmonizing revisions to part 352 and § 201.327. These changes remove certain provisions from part 352 that were superseded by the 2011 L&E Final Rule and, where applicable, replace them with appropriate cross references to the applicable testing and labeling provisions in § 201.327, as we propose to amend these regulations in this document. We also have made minor revisions in parts 347, 352 and § 201.327 to improve readability and to correct certain typographical errors and erroneous internal cross references.Start Printed Page 6249
We are also proposing revisions to certain provisions describing requirements for products containing both sunscreen active ingredients and skin protectant active ingredients to avoid duplication between § 201.327 and part 352 and to harmonize the requirements set forth in those provisions. As in the past, the proposed sunscreen monograph would include conditions under which a single product could include certain sunscreen active ingredients as well as certain ingredients determined to be GRASE for use in skin protectants under part 347 (see proposed § 352.20(b), as well as current § 347.20(e) (21 CFR 347.20(e)). Current § 201.327(h) allows for such products to combine certain labeling statements applicable to each ingredient in the product to eliminate duplicative words or phrases. The stayed provisions of part 352 contain similar allowances for products that contain both sunscreen and skin protectant active ingredients, but also outline more detailed requirements for presenting such a product's statement of identity, indications, warnings, and directions. We propose to relocate the labeling requirements for such products from § 352.60 to § 201.327(h), thereby consolidating labeling conditions for these products in one section of the regulations. We also propose to retain compliance with these labeling provisions as a monograph condition for sunscreen/skin protectant products under both parts 352 (the sunscreen monograph) and 347 (the skin protectant monograph) by incorporating cross references to § 201.327(h) in § 352.20(b)(4), and § 352.60, and incorporating cross references to §§ 352.20 and 352.60 in §§ 347.20(e), and 347.60.
Additionally, we propose to consolidate under new § 310.549 (21 CFR 310.549) certain properties that render an OTC drug product offered for use as sunscreen a new drug for which an approved NDA is required prior to marketing. Section 310.545 (21 CFR 310.545) currently contains several such provisions addressing specific ingredients and efficacy claims. We propose to relocate these provisions from § 310.545 to § 310.549. In addition, in the interest of completeness, we are clarifying in § 310.549 that labeling a product with claims that it decreases the risk of skin cancer or early skin aging caused by the sun if that product has an SPF of less than 15 when tested in accordance with § 201.327(i) and/or does not pass the broad spectrum test in § 201.327(j) renders the product a new drug.
Finally, we propose to add to § 310.549 new characteristics that would render a product a new drug. These characteristics include: (1) Containing the ingredients we propose to classify as categories II and III (see sections VIII.B-C); (2) being labeled, represented, or promoted for use as a combined sunscreen-insect repellant (see section IX.E); (3) failing to comply with provisions relating to maximum SPF values and broad spectrum requirements (see section IX.B); and (4) failing to conform to certain other sunscreen formulation and dosage form conditions (see sections IX.A and D).
XI. Comment Period
We are providing a comment period of 90 days (see DATES). FDA will also consider requests to defer further rulemaking with respect to a specific sunscreen active ingredient to allow the submission of new safety and/or effectiveness data to the record if such requests are submitted to the docket within the initial 90-day comment period. FDA will review all data and information submitted to the record in conjunction with all timely and complete requests to extend. In assessing whether to extend the comment period to allow for additional time for studies to generate new data and information, FDA will consider the data already in the docket along with any information that is provided in any requests to extend. FDA will determine whether the sum of the data, if timely submitted, is likely to be adequate to provide all the data that are necessary to make a determination of general recognition of safety and effectiveness.
XII. Proposed Effective/Compliance Dates
The proposed effective date of final regulations resulting from the proposals described in this rulemaking is November 26, 2019 (see FD&C Act section 586E). We recognize that industry will need time after publication of any final regulations to comply with their provisions. To allow for orderly implementation of final regulations and help assure continued product availability to consumers, we would not expect full compliance with such final regulations for units of sunscreen product initially introduced or initially delivered for introduction into interstate commerce, until 1 year after the effective date of the final rule. We also would not expect full compliance, even after that date, for units of product that were initially introduced or initially delivered for introduction into interstate commerce before that date, such as those remaining in retail outlets. Our current thinking on implementation is informed in part by our understanding there are no currently marketed sunscreen products that contain the active ingredients we propose here as Category II. We solicit comment on this proposed approach.
XIII. Preliminary Economic Analysis of Impacts
A. Introduction
We have examined the impacts of the proposed rule under Executive Order 12866, Executive Order 13563, Executive Order 13771, the Regulatory Flexibility Act (5 U.S.C. 601-612), and the Unfunded Mandates Reform Act of 1995 (Pub. L. 104-4). Executive Orders 12866 and 13563 direct us to assess all costs and benefits of available regulatory alternatives and, when regulation is necessary, to select regulatory approaches that maximize net benefits (including potential economic, environmental, public health and safety, and other advantages; distributive impacts; and equity). Executive Order 13771 requires that the costs associated with significant new regulations “shall, to the extent permitted by law, be offset by the elimination of existing costs associated with at least two prior regulations.” We believe that this proposed rule is a significant regulatory action as defined by Executive Order 12866.
The Regulatory Flexibility Act requires us to analyze regulatory options that would minimize any significant impact of a rule on small entities. Because many sunscreen manufacturers are small entities and the one-time costs of the proposed rule represent a significant fraction of annual revenue to sunscreen manufacturers, we find that the proposed rule will have a significant economic impact on a substantial number of small entities.
The Unfunded Mandates Reform Act of 1995 (section 202(a)) requires us to prepare a written statement, which includes an assessment of anticipated costs and benefits, before proposing “any rule that includes any Federal mandate that may result in the expenditure by State, local, and tribal governments, in the aggregate, or by the private sector, of $100,000,000 or more (adjusted annually for inflation) in any one year.” The current threshold after adjustment for inflation is $150 million, using the most current (2017) Implicit Price Deflator for the Gross Domestic Product. This proposed rule would result in an expenditure in any year that meets or exceeds this amount.Start Printed Page 6250
We have developed a comprehensive preliminary regulatory impact analysis that assesses the impacts of the proposed rule. We present a summary of this analysis below.
B. Summary of Costs and Benefits
If finalized, the proposed rule would update and make effective regulations to ensure the safety and effectiveness of sunscreen products marketed under the OTC drug monograph. The rule would update sunscreen product labeling standards, address the safety of sunscreen active ingredients, revise and clarify our expectations for testing and recordkeeping by entities that conduct sunscreen testing, and address other sunscreen safety or efficacy concerns, like combination sunscreen-insect repellents and alternative dosage forms.
Consumers would benefit from less exposure to sunscreen products containing active ingredients about which safety questions remain, less exposure to sunscreen products labeled with potentially misleading sun protection information, increased consumption of products with better UVA protection, less exposure to flammable spray sunscreens, and less exposure to spray and powder sunscreen products posing inhalation risks. Consumers would also experience transaction cost savings. The costs of the rule to sunscreen manufacturers include administrative costs, costs to fill data gaps for active ingredients and powder dosage forms, product formulation testing costs, and costs to reformulate and relabel sunscreen products. Finally, testing entities would incur recordkeeping costs if they do not already maintain adequate records of testing equipment, methods, and observations in final formulation testing.
Table 7 summarizes the costs and benefits of the proposed rule, if finalized. The annualized benefits of the proposed rule, if finalized, would range from $0.00 million to $3.72 million at a 7 percent discount rate and from $0.00 million to $3.62 million at a 3 percent discount rate. Our primary estimate of annualized benefits would equal $0.91 million at a 7 percent discount rate and $0.88 million at a 3 percent discount rate. The annualized costs of the proposed rule, if finalized, would range from $15.57 million to $75.84 million at a 7 percent discount rate and from $12.40 million to $60.42 million at a 3 percent discount rate. Our primary estimate of annualized costs would be $47.55 million at a 7 percent discount rate and $37.79 million at a 3 percent discount rate.[52]
Table 7—Summary of Benefits, Costs, and Distributional Effects of the Proposed Rule
Category Primary estimate Low estimate High estimate Units Notes Year dollars Discount rate (%) Period covered (years) Benefits: Annualized Monetized ($m/year) $0.91 0.88 $0.00 0.00 $3.72 3.62 2017 2017 7 3 20 20 Annualized Quantified (mil oz/year) 1 201.79 98.16 286.26 Increased use of products with improved UVA protection. Annualized Quantified (mil oz/year) 2 51.42 19.43 83.41 Less exposure to sunscreens containing active ingredients about which safety questions remain. Annualized Quantified (mil oz/year) 3 161.04 159.88 162.20 Less exposure to sunscreens with potentially misleading sun protection information. Annualized Quantified (mil oz/year) 4 386.44 384.86 388.02 Less exposure to spray and powder sunscreens posing inhalation risks. Qualitative Quicker responses to adverse events, improved inspections, and better protection of human subjects. Potential transaction cost savings related to changes in the effort required to choose a sunscreen. Costs: Annualized Monetized ($m/year) 47.55 37.79 15.57 12.40 75.84 60.42 2017 2017 7 3 20 20 Annualized Quantified Qualitative Recordkeeping costs to testing entities that do not already maintain adequate records. Transfers: Federal Annualized Monetized ($m/year) Start Printed Page 6251 From: To: Other Annualized Monetized ($m/year) From: To: Effects: State, Local, or Tribal Government: None. Small Business: Some small businesses could exit the sunscreen market by discontinuing their products or going out of business. Wages: None. Growth: None. 1 Values represent the 2016 consumption of sunscreens that would provide improved UVA protection under the proposed rule. 2 Value represent the 2016 consumption of sunscreens that contain active ingredients about which safety questions remain. 3 Values represent the 2016 consumption of sunscreens with potentially misleading sun protection information. 4 Values represent the 2016 consumption of potentially inhalable spray sunscreens and powder sunscreens. Table 8 shows the Executive Order 13771 summary over an infinite time horizon. In this analysis we assume that the costs and cost savings of the rule would end after 20 years. We estimate that this rule generates $29.85 million in net annualized costs, discounted at 7 percent, over a perpetual time horizon. Based on these costs, this proposed rule would be considered a regulatory action under E.O. 13771.
Table 8—E.O. 13771 Summary Table
[In $ millions 2016 dollars, over an infinite time horizon) 1
Primary estimate (7%) Lower bound (7%) Upper bound (7%) Primary estimate (3%) Lower bound (3%) Upper bound (3%) Present Value of Costs $456.33 $149.22 $730.46 $618.16 $201.53 $1,002.22 Present Value of Cost Savings 0.00 0.00 0.00 0.00 0.00 0.00 Present Value of Net Costs 456.33 149.22 730.46 618.16 201.53 1,002.22 Annualized Costs 29.85 9.76 47.79 40.44 13.18 65.57 Annualized Cost Savings 0.00 0.00 0.00 0.00 0.00 0.00 Annualized Net Costs 29.85 9.76 47.79 40.44 13.18 65.57 1 We assume that the benefits and costs of the proposed rule would diminish after 20 years. Negative values denoted in parentheses. We have developed a comprehensive Economic Analysis of Impacts that assesses the impacts of the proposed rule. The full preliminary analysis of economic impacts is available in the docket for this proposed rule (Ref. 63) and at https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/EconomicAnalyses/default.htm.
XIV. Analysis of Environmental Impact
We have determined under 21 CFR 25.31(c) that this action is of a type that does not individually or cumulatively have a significant effect on the human environment. Therefore, neither an environmental assessment nor an environmental impact statement is required.
XV. Paperwork Reduction Act of 1995
This proposed rule contains information collection provisions that are subject to review by OMB under the Paperwork Reduction Act of 1995 (PRA) (44 U.S.C. 3501-3520). A description of these provisions is given in the Description section of this document with an estimate of the annual reporting, recordkeeping, and third-party disclosure burden. Included in the estimate is the time for reviewing instructions, searching existing data sources, gathering and maintaining the data needed, and completing and reviewing each collection of information.
FDA invites comments on these topics: (1) Whether the proposed collection of information is necessary for the proper performance of FDA's functions, including whether the information will have practical utility; (2) the accuracy of FDA's estimate of the burden of the proposed collection of information, including the validity of the methodology and assumptions used; (3) ways to enhance the quality, utility, and clarity of the information to be collected; and (4) ways to minimize the burden of the collection of information on respondents, including through the use of automated collection techniques, when appropriate, and other forms of information technology.
Title: Sunscreen Drug Products for OTC Human Use.
Description: The proposed rule would amend FDA's current sunscreen labeling regulation (§ 201.327) and sunscreen products monograph (part 352) regarding product labeling, testing, and recordkeeping requirements. We note that existing regulations (e.g., current § 201.327) already require SPF testing and labeling. The information collections associated with current testing, labeling, and recordkeeping requirements have previously been approved in accordance with the PRA under OMB control numbers 0910-0139, 0910-0717, and 0910-0755. For more information about current regulations and their history, see the Background and Scope sections of the Start Printed Page 6252proposed rule (sections III and IV, respectively). The proposed rule would also amend parts 310 and 347.
While the proposed provisions are broadly consistent with current best practices for testing conducted in human subjects and are not expected to require significant changes by reputable and experienced testing establishments, the proposed rule clarifies and confirms the application of existing requirements to sunscreens and adds certain new requirements, particularly for labeling and recordkeeping. The purpose of these changes is to help ensure that sunscreen testing is conducted and documented in a way that verifiably provides for protection of human subjects and increases the reliability of the testing data that underlies sunscreen labeling, and to update the labeling requirements.
Description of Respondents: Affected entities include: (1) “responsible persons,” as defined in proposed § 201.327(a); (2) entities to which the responsible person transfers its obligations as permitted under proposed § 201.327(a)(1) (e.g., contract manufacturers, contract testing entities, contract research organizations); and (3) clinical investigators conducting the testing (the investigator(s) required to submit investigator statements and other materials to the responsible person).
FDA estimates that up to 772 entities could meet the proposed definition of responsible person (equivalent to “brands” in the economic analysis found in section XIII, Preliminary Analysis of Economic Impacts). The estimate of 772 entities also includes nearly all entities to which a responsible person might transfer its obligations (“transferees”), such as contract manufacturers, contract repackagers, contract distributors. For example, a manufacturer may be a responsible person for one brand and a contract manufacturer for another. However, in addition to the 772 entities and potential transferees already described, we estimate that there are approximately 10 U.S.-based contract testing entities used by multiple responsible persons to conduct sunscreen testing (e.g., contract laboratories and contract research organizations). These 10 potential transferees are not included in the 772 figure. Thus, for certain information collections, the estimated respondent number may be 782. We note that this estimate does not include non-U.S.-based contract testing entities.
In addition to the 10 contract testing entities, FDA estimates that approximately 10 of the estimated 772 responsible persons conduct their own SPF or broad spectrum testing. Thus, we estimate that there are approximately 20 entities that conduct covered sunscreen testing; we estimate these entities have approximately 20 lead clinical investigators to whom certain information collection obligations (e.g., reporting) may apply.
A. Labeling for Sunscreen Products and Associated Clinical Testing
The proposed rule includes third-party disclosure obligations for responsible persons. The provisions may also apply to entities to which the responsible persons transfer their responsibilities under section 201.327(a)(1) (“transferees”), depending on the scope of transferred obligations. There are labeling-related information collections (requirements include certain information on product labels) and a related testing burden (requirements for certain clinical testing to determine and support labeling information).
1. Labeling-Related Information Collection and Burden
Proposed § 201.327(b) and § 201.327(h)(1) amend certain labeling requirements applicable to the PDP. Among other things, proposed § 201.327(b) sets forth labeling requirements for the statement of identity and SPF value claims discussed in this section. Proposed § 201.327(h)(1) applies to the statement of identity for sunscreen products that also contain skin protectant active ingredients. Proposed § 352.50 requires that the PDP labeling comply with the requirements of § 201.327(b). The SPF value statements set forth in proposed § 201.327(b) and referenced in proposed § 352.50 are based on the results of the testing required in proposed § 201.327(i) and proposed part 352 (§ 352.70).
a. Statement of identity. Proposed § 201.327(b)(1) requires that sunscreen drug products bear a statement of identity consisting of the name of each sunscreen active ingredient listed in alphabetical order, followed by “Sunscreen” and “[Dosage form]” (e.g., “Lotion”, “Spray”). Proposed § 352.52(a) requires the labeling to contain a statement of identity in accordance with § 201.327(b).
Proposed § 201.327(h)(1) applies to sunscreen drug products that also contain skin protectant active ingredients; it requires that the product bear a statement of identity consisting of the name of all sunscreen and skin protectant active ingredients in alphabetical order, followed by “Sunscreen/Skin Protectant” and “[Dosage form],” presented in accordance with § 201.327(b)(1)(ii). Proposed § 352.60(a) requires that the product bear a statement of identity as set forth in § 201.327(h)(1). Proposed § 352.20(b)(4) requires that the product must be labeled in accordance with §§ 201.327(h) and 352.60. Proposed § 347.60(a)(3) requires that the labeling of the product bear the statement of identity set forth in proposed § 352.60(a).
We note that current regulations already include a requirement that OTC products bear a statement of identity (see § 201.66). This proposed rule would set forth the specific requirements just described for sunscreen drug products and sunscreen drug products that also contain skin protectant active ingredients. We believe this analysis reflects the additional burden beyond current statement of identity requirements.
b. SPF value. Proposed § 201.327(b)(2) requires, among other things, that the labeling display certain statements regarding the product's SPF value; the statements must be supported by the testing required by proposed § 201.327(i) and referenced in proposed § 352.70. As previously noted, certain SPF testing and labeling is already required under current regulations. This analysis reflects the estimated additional burden of the proposed changes to SPF testing and labeling requirements.
c. Burden for proposed statement of identity and SPF value information collections. The estimated burden for the statement of identity and SPF value information collections just described is provided in table 11 (Estimated Annual Third-Party Disclosure Burden). For currently marketed OTC sunscreen products, FDA believes that responsible persons need only complete the testing (or reanalyze existing testing data) and relabel the product as required by the rule one time, and may then continue to utilize the resultant labeling going forward without additional burden. We estimate that 772 respondents would need to complete this relabeling and related testing (if not already done) or reanalysis of existing test results one time for up to 4,078 total products. In addition, there may be new products introduced each year. We estimate that as many as 1,500 new OTC sunscreen product stock keeping units (SKUs) may be introduced each year by up to 772 respondents. These new products must be tested and labeled with the SPF value and broad spectrum results determined in the tests. We estimate that the 1,500 new sunscreen SKUs represent 975 new formulations.Start Printed Page 6253
Table 11, row 1 provides FDA's estimate that 772 respondents will need to create PDP labeling for currently marketed sunscreen formulations in accordance with the statement of identity and SPF value requirements of proposed § 201.327(b)(1), (b)(2), and (h)(1). This would be a one-time burden, and FDA estimates 5.2824 responses per respondent for a total of 4,078 responses. FDA estimates a burden of 0.5 hours per response. We estimate the total burden of this recordkeeping to be 2,039 hours.
Table 11, row 2 provides FDA's estimate that up to 772 respondents will need to create PDP labeling for new formulations each year in accordance with the statement of identity and SPF value requirements of proposed § 201.327(b)(1), (b)(2), and (h)(1). FDA estimates 1.943 responses per respondent for a total of 1,500 responses. FDA estimates a burden of 0.5 hours per response. We estimate the total burden of this recordkeeping to be 750 hours.
Table 11, row 3 provides FDA's estimate that 20 respondents will conduct SPF testing in accordance with § 201.327(i) (to determine the SPF value required by § 201.327(b)(2)) for currently marketed sunscreen formulations, if this has not already been done. This would be a one-time burden. The estimated number of respondents reflects FDA's assumption based on its knowledge of the existing market that, of the 772 responsible persons, approximately 10 will conduct their own final formulation testing under § 201.327(i), while most will delegate the responsibility for conducting final formulation testing to the approximately 10 independent testing entities that FDA believes conduct most final formulation testing. FDA estimates 111 responses per respondent for a total of 2,220 responses. FDA estimates a burden of 24 hours per response. We estimate the total burden to be 53,280 hours.
Table 11, row 4 provides FDA's estimate that 20 respondents will conduct SPF testing in accordance with § 201.327(i) (to determine the SPF value required by § 201.327(b)(2)) for new sunscreen formulations. FDA estimates 48.75 responses per respondent for a total of 975 responses. FDA estimates a burden of 24 hours per response. We estimate the total burden to be 23,400 hours.
Regarding proposed § 352.70, because that section does not add any additional labeling or testing-related information collections not already addressed elsewhere (it incorporates the proposed SPF testing requirements as a condition of the part 352 monograph), there is no additional burden.
2. Clinical Testing-Related Information Collection and Burden
Proposed § 201.327(i) contains requirements for clinical testing of SPF values for inclusion on sunscreen product labeling. As previously noted, current regulations already require SPF testing. While FDA expects that SPF testing and some of the proposed recordkeeping is already being done, the proposed changes are intended to clarify existing requirements applicable to sunscreen drug products and set forth certain new requirements intended to improve the reliability of SPF testing and ensure the protection of human subjects. Proposed § 352.70 references the § 201.327(i) testing requirements and makes the referenced testing requirements part of the monograph conditions of use.
Across disciplines, testing involving human subjects is ordinarily conducted under IRB oversight as a means of ensuring that adequate human subject protections are provided and to ensure the integrity of study design and execution. Thus, in this proposed regulation, FDA is proposing to apply certain human subject protection requirements to sunscreens, with the aim of having a framework similar to that used in the IND context, but tailored to sunscreen testing.
Information collections related to proposed § 201.327(i) are addressed in detail in the sections that follow. Regarding proposed § 352.70, as in the previous section, because the proposed change does not add any additional labeling or testing-related information collections not already addressed elsewhere (it cross-references the proposed § 201.327(i) testing requirements as a condition of the part 352 monograph), there is no additional burden.
a. Investigator statements and notifications. Proposed § 201.327(i), among other things, requires responsible persons to obtain a signed investigator statement and an investigator CV, and to provide certain notifications (e.g., notification of adverse drug experiences). These may result in third-party disclosure or reporting requirements for responsible persons (and entities to which they have transferred relevant obligations) as well as for clinical investigators. As noted above, our experience leads us to believe that most responsible persons will transfer their obligations under § 201.327(i) to the approximately 20 entities that currently conduct clinical SPF testing. This assumption is reflected in the estimates regarding the number of respondents below.
b. Investigator statements, CVs, and related burden. Proposed § 201.327(i)(1)(i) requires responsible persons to, among other things, obtain a signed investigator statement from each investigator. Proposed § 201.327(i)(1)(iv)(B) requires responsible persons to obtain a signed investigator statement and CV. In FDA's experience, investigators for SPF testing are most often employed by the testing entities, and we therefore believe this is a recordkeeping requirement rather than a third-party reporting requirement. We request comment on this assumption. As noted above, we estimate that responsible persons will typically delegate this obligation to the approximately 20 entities conducting final formulation testing. We estimate that each testing entity employs one clinical investigator to run the SPF testing they conduct. One investigator may run multiple SPF tests, and so long as the responsible person (or testing entity) has the investigator statement and CV on file for each investigator, there need not be a separate copy for each investigation.
Table 10, row 1 provides FDA's estimate that approximately 20 respondents will need to obtain and keep a signed investigator statement and CV in accordance with § 201.327(i)(1)(i) and (i)(1)(iv)(B). FDA estimates 2 responses per respondent (1 CV and 1 investigator statement) for a total of 40 annual responses. FDA estimates a burden of 0.6 hours per response. We estimate the total burden of this recordkeeping to be 24 hours.
c. Notifications and related burden. Proposed § 201.327(i)(1)(i) requires responsible persons to, among other things, ensure that FDA and all participating investigators are promptly informed of significant new adverse effects or risks with respect to the drug. Proposed § 201.327(i)(1)(v) requires responsible persons to keep each participating investigator informed of new observations about the drug, particularly with respect to adverse effects and safe use. As mentioned above, like other obligations associated with testing under proposed § 201.327(i), we anticipate that this obligation will be delegated in most instances to the approximately 20 entities that currently conduct SPF testing on behalf of responsible persons.
Table 9, row 1 provides FDA's estimate that approximately 20 respondents will need to inform FDA and participating investigators of significant new adverse effects or risks in accordance with § 201.327(i)(1)(i) and Start Printed Page 6254of new safety and other observations in accordance with § 201.327(i)(1)(v). FDA estimates up to 40 responses per respondent for a total of up to 800 annual responses. FDA estimates a burden of 0.5 hours per response. We estimate the total burden of this recordkeeping to be 400 hours.
d. Informed consent, IRB review, and related burden. Proposed § 201.327(i)(1)(ii) requires responsible persons to obtain informed consent, as defined in part 50, before clinical final formulation testing and proposed § 201.327(i)(1)(iii) requires that clinical testing under § 201.327(i) be reviewed and approved by an IRB meeting the requirements of part 56. These two proposed provisions make clear that FDA's regulations governing informed consent (part 50) and IRB approval of research (part 56) apply to clinical final formulation testing conducted pursuant to § 201.327(i).
Regarding proposed § 201.327(i)(1)(ii) and (iii), the information collections associated with FDA's regulations governing informed consent (part 50) and IRB approval of research (part 56) have previously been approved in accordance with the PRA under OMB control number 0910-0755. FDA does not expect that proposed § 201.327(i)(1)(ii) or (iii) would affect the number of recordkeepers, records, reports, or associated burdens included in the existing approval (0910-0755), but we invite stakeholders to comment if they have a different view.
Proposed § 201.327(i)(1)(vii) requires investigators to provide safety reports and a final study report to the responsible person. Although investigators are often employees of testing entities, we are basing our estimate on our assumption the respondents in this case are the investigators themselves because of the framing of the duty proposed by the regulation.
Table 9, row 2 provides FDA's estimate that up to 20 respondents will need to provide safety reports in accordance with § 201.327(i)(1)(vii)(A). FDA estimates 24.4 responses per respondent for a total of 488 annual responses. FDA estimates a burden of 0.5 hours per response. We estimate the total burden of this recordkeeping to be 244 hours.
Table 9, row 3 provides FDA's estimate that up to 20 respondents will need to provide a final report in accordance with § 201.327(i)(1)(vii)(B). This will occur one time per study, with each of the 20 investigators conducting multiple studies per year. FDA estimates 48.75 responses per respondent for a total of 975 annual responses. FDA estimates a burden of 3 hours per response. We estimate the total burden of this recordkeeping to be 2,925 hours.
Proposed § 352.40(i)(1) references limitations on particle size for sunscreens in a spray dosage form. Proposed § 352.40(i)(2) and (3) proposes limitations on flammability and drying time for spray sunscreen formulations. These proposed sections (§ 352.40(i)(1) through (3)) make the referenced limitations on flammability and particle size requirements part of the monograph conditions of use. Proposed § 352.40(i)(5) states that applicable requirements for particle size, flammability, and drying time for spray sunscreens must be verified through batch and lot testing as part of CGMP compliance under part 211. Entities conducting testing required by these sections must also comply with associated recordkeeping requirements, including those set forth in parts 210 and 211.
The recordkeeping associated with ensuring compliance with § 352.40(i)(5) (batch and lot testing to ensure compliance with particle size, flammability, and drying time limitations) is considered to be part of the manufacturers' CGMP requirements under parts 210 and 211 (OMB control number 0910-0139). While FDA believes that sunscreen manufacturers are already included among the respondents counted for that collection, and that many of those manufacturers who have spray dosage products may already be conducting flammability and drying time testing (e.g., many are including flammability statements and information about drying time in current product labeling), the proposed inclusion of these requirements in the sunscreen regulations is new. The proposed rule specifies the particle size, flammability, and drying time limitations that would be required for sunscreens in spray dosage forms to be GRASE under the monograph. The proposed rule also specifies that compliance with these limitations must be verified through batch and lot testing. While this greater specificity as to required testing might have a marginal effect on the burden associated with recordkeeping for manufacturing facilities that are not already conducting such testing, FDA believes that the total change would be minimal in light of the total recordkeeping burden under parts 210 and 211, which is estimated across thousands of manufacturers of a wide variety of drugs. We request comment on these assumptions. If FDA determines that the assumptions are incorrect, then, concurrent with publication of the final rule, FDA plans to amend its approved information collection 0910-0139, if necessary, to adjust the respective burden estimate(s) to account for any change. We request comment on the accuracy of our assumptions and the resulting burden estimate.
B. Regulatory Status of Testing Entities
Proposed § 201.327(k) clarifies the regulatory status of final formulation testing, including that final formulation testing conducted pursuant to § 201.327 constitutes “manufacture” of a drug. As such, this testing must be conducted in an establishment registered in accordance with part 207 and section 510 of the FD&C Act, and entities conducting final formulation testing required by this section must comply with CGMP and associated recordkeeping requirements, including those set forth in § 201.327(l) and in parts 210 and 211. As this provision is intended only to clarify an existing requirement, it does not create a new information collection.
Entities covered by this provision are already included in the burden estimates for the information collections associated with registration and listing requirements. Recordkeeping obligations related to registration and listing under part 207 and section 510 of the FD&C Act are part of FDA's approved information collection for part 207 (OMB control number 0910-0829). CGMP recordkeeping obligations are part of FDA's approved information collection for part 211 (OMB control number 0910-0139).
C. Generating and Maintaining Records of SPF and Broad Spectrum Testing
FDA is proposing specific recordkeeping requirements for SPF and broad spectrum testing to enable FDA to better monitor responsible persons' compliance with the requirements of § 201.327. Recordkeeping is essential for FDA to evaluate whether required testing of final formulations is being conducted properly (both as to human subject protection and as to study design) and to enable the Agency to investigate postmarketing product failures or adverse events. Appropriate recordkeeping also enables FDA to conduct better and more efficient inspections of entities conducting final formulation testing. The proposed recordkeeping requirements are in alignment with the records required for other types of manufacturing under CGMPs as set forth in parts 210 and 211.
Failure to maintain adequate records of testing equipment, methods, and observations can raise broad questions Start Printed Page 6255about the reliability of final formulation testing. In FDA's experience, there has been a lack of uniformity in testing entities' approaches to recordkeeping for final formulation testing, raising concerns about the Agency's ability to assess the reliability of the results of final formulation testing. The proposed regulation would address these concerns, clarify FDA's expectations, and align the regulation with current best practices.
a. Potential transfer of obligations. Proposed § 201.327(a)(1) permits a responsible person (defined in § 201.327(a)) to transfer some or all of its obligations to another entity (a “transferee”), except for obligations with respect to recordkeeping under § 201.327(l). We note that this could create some situations in which both the responsible person and the transferee would be required to comply with applicable recordkeeping requirements. The proposed provision would also require a written record of the transfer of obligations to be maintained by both parties to the transfer.
Regarding the record of an obligation transfer, FDA believes that it is usual and customary business practice for a written record of a transfer of obligations to be maintained by both parties to the transfer. FDA does not believe this requirement would incur any additional recordkeeping burden and believes it would meet the exception at 5 CFR 1320.3(b)(2). Regarding the potential for some recordkeeping obligations to fall on both responsible persons and transferees, although proposed § 201.327(a)(1) does not itself impose a specific requirement to generate records, it does create the potential for some recordkeeping obligations to fall on both responsible persons and transferees. In particular, if a responsible person has delegated all other responsibilities under § 201.327(i) and (j), they would nonetheless need to maintain a copy of the records of final formulation testing required by § 201.327(l)(2) and (3). We have included the burden associated with keeping this copy in our assumption that there are 782 respondents for recordkeeping obligations as described below (20 entities that conduct testing, 10 of whom are also responsible persons, plus 762 responsible persons that delegate their responsibility for conducting testing (e.g., to one of the 10 independent testing entities that are not themselves responsible persons)). We invite comment on whether our estimates properly reflect the recordkeeping obligations.
b. Maintenance records and related burden. Proposed § 201.327(l)(1) addresses maintenance records. The proposed rule clarifies that, as manufacturing, final formulation testing must comply with CGMPs, and, accordingly, records documenting proper maintenance of equipment used in final formulation testing must be generated and maintained by testing entities, consistent with existing obligations in part 211.
Regarding proposed § 201.327(l)(1), the existing maintenance record obligations are part of FDA's approved information collection for part 211 (OMB control number 0910-0139), and FDA believes that most of the respondents for this collection of information (the approximately 20 entities we believe are conducting final formulation testing) are already included among the recordkeepers counted for that collection. The proposed rule provides greater specificity regarding what information should be included in maintenance records maintained by facilities conducting final formulation testing. While this greater specificity might have a marginal effect on the burden associated with recordkeeping for these facilities, and the number of respondents for this requirement may need to be increased by 10 (to reflect contract testing entities that may not be currently registered as manufacturers), FDA believes that the total change would be minimal in light of the total recordkeeping burden under parts 210 and 211, which is estimated across thousands of manufacturers of a wide variety of drugs. We request comment on these assumptions. If FDA determines that the assumptions are incorrect, then, concurrent with publication of the final rule, FDA plans to amend its approved information collection under OMB control number 0910-0139 as necessary to adjust the respective burden estimate(s) in order to account for any change.
c. SPF testing records and related burden. Proposed § 201.327(l)(2) addresses SPF testing records and requires that respondents keep records related to the identification of the entity conducting the testing, the formulation being tested, equipment used, investigators, SPF standards, specific subject and test result data, and records demonstrating compliance with § 201.327(i)(1) governing the establishment of adequate clinical testing procedures and conditions. This is important because failure of testing entities to keep adequate records to support final formulation testing may leave FDA unable to verify the reliability of the results of SPF testing. Because one testing entity may conduct final formulation testing on behalf of multiple responsible persons, an error at one testing entity may affect data across multiple clinical SPF testing studies for multiple different final formulations that are ultimately sold under different labels.
In particular, proposed § 201.327(l)(2) requires that, in addition to any records required to be kept pursuant to parts 210 and 211, records of SPF testing must include: (1) Identification of the testing entity; (2) the product identifier and expected SPF; (3) characterization of the SPF standard sunscreen required by proposed § 201.327(i)(3) (lot number, manufacturing date, and results of high performance liquid chromatography (HPLC) SPF standard assay); (4) documentation linking any blinded samples with the product lot number and formulation tested; (5) specific testing records for each human subject (identification of the UV source used for testing and various specific test results and the individual(s) who determined the values); (6) the mean and standard deviation from SPFi values, standard error and determined SPF value derived as set forth in proposed § 201.327(i)(7); (7) records for water resistance testing of pool temperature, air temperature, and relative humidity as required by proposed § 201.327(i)(8); and (8) records demonstrating compliance with proposed § 201.327(i)(1) requirements for adequate clinical testing procedures and conditions (e.g., individual case histories and documentation of IRB review).
Table 10, row 2 provides FDA's estimate that approximately 20 respondents will need to generate SPF testing records in accordance with proposed § 201.327(l)(2) for existing products that will be reformulated. FDA estimates 85.5 records per recordkeeper for a total of 1,710 records. This is a one-time burden. FDA estimates a burden of 24 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 41,040 hours.
Table 10, row 3 provides FDA's estimate that up to 20 respondents will need to generate SPF testing records in accordance with proposed § 201.327(l)(2) for new formulations. FDA estimates 48.75 records per recordkeeper for a total of 975 records. FDA estimates a burden of 24 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 23,400.
Table 10, row 4 provides FDA's estimate that up to 782 respondents will need to keep SPF testing records in accordance with proposed § 201.327(l)(2) for existing products that will be reformulated. This is a one-time Start Printed Page 6256burden. FDA estimates 2.1867 records per recordkeeper for a total of 1,710 records. FDA estimates a burden of 0.33 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 564.3 hours.
Table 10, row 5 provides FDA's estimate that up to 782 respondents will need to keep SPF testing records in accordance with proposed § 201.327(l)(2) for new formulations. FDA estimates 1.2468 records per recordkeeper for a total of 975 records. FDA estimates a burden of 0.33 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 321.75 hours.
With regard to the testing-related estimates, we note that the requirements for obtaining certain medical history information from test subjects are not considered collections of information because information collected from subjects of clinical testing does not constitute information under 5 CFR 1320.3(h)(5), and that the referenced informed consent and IRB requirements under parts 50 and 56 are covered by existing approvals, as previously discussed.
d. Broad spectrum testing records and related burden. Proposed § 201.327(l)(3) addresses broad spectrum testing records. The proposed rule requires records related to the identification of the entity conducting the testing, the formulation being tested, equipment used, investigators, UV standards, sunscreen application, and specific test result data. This is important because failure of testing entities to keep adequate records to support broad spectrum testing may leave FDA unable to verify the reliability of testing results. Failure at one testing entity may affect data across multiple broad spectrum testing studies for multiple different final formulations that are ultimately sold under different labels.
In particular, proposed § 201.327(l)(3) requires that records of broad spectrum testing must include: (1) Identification of the testing entity; (2) records of sample information (product identifier and expected SPF, master key for blinded samples, sample number and identifier code, polymethylmethacrylate (PMMA) plate surface topography measurement, and sample holder orientation); (3) identification of each UV source used for sunscreen product pre-irradiation; (4) records of sunscreen product application (sample weights, equipment identification); (5) measurements required by proposed § 201.327(j)(4) to (6)); (6) records of critical wavelength and UVA1/UV ratio values; (7) for each sample: The identity of the individual(s) conducting specific testing steps; and (8) test dates for the broad spectrum test conducted pursuant to § 201.327(j), and sample report forms and supporting data.
Table 10, row 6 provides FDA's estimate that approximately 20 respondents will need to generate broad spectrum testing records in accordance with proposed § 201.327(l)(3) for existing products. As with records of SPF testing, this number of respondents reflects FDA's assumption that most responsible persons will delegate responsibility for conducting testing under § 201.327(j) to the approximately 20 testing entities. FDA estimates 203.9 records per recordkeeper for a total of 4,078 records. This is a one-time burden. FDA estimates a burden of 1.5 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 6,117 hours.
Table 10, row 7 provides FDA's estimate that up to 20 respondents will need to generate broad spectrum testing records in accordance with proposed § 201.327(l)(3) for new formulations. FDA estimates 48.75 records per recordkeeper for a total of 975 records. FDA estimates a burden of 1.5 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 1,462.5 hours.
Table 10, row 8 provides FDA's estimate that up to 782 respondents will need to keep broad spectrum testing records in accordance with proposed § 201.327(l)(3) for existing products. This is a one-time burden. FDA estimates 5.215 records per recordkeeper for a total of 4,078 records. FDA estimates a burden of 0.17 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 693.3 hours.
Table 10, row 9 provides FDA's estimate that up to 782 respondents will need to keep broad spectrum testing records in accordance with proposed § 201.327(l)(3) for new formulations. FDA estimates 1.2468 records per recordkeeper for a total of 975 records. FDA estimates a burden of 0.17 hours per recordkeeping. We estimate the total burden of this recordkeeping to be 165.75 hours.
The recordkeeping burden is estimated as described in the tables at the end of the PRA discussion.
With the exceptions noted above, we conclude that the other provisions of this rule are not subject to OMB review under the PRA.
The proposed changes to part 310 do not include any collections of information subject to the PRA.
The remaining sections of part 347 do not include any collections of information not already addressed in this analysis.
Section 201.327 and the remaining sections of part 352 either do not contain an information collection subject to PRA, or contain specific labeling information, including directions and warnings, which are a “public disclosure of information originally supplied by the Federal Government to the recipient for the purpose of disclosure to the public” (5 CFR 1320.3(c)(2)) and, therefore, are not collections of information.
FDA estimates the burden of this information collection as follows:
Start Printed Page 6257Table 9—Estimated Annual Reporting Burden 1
Activity and 21 CFR section Number of respondents Number of responses per respondent Total annual responses Average burden per response Total hours Inform FDA and investigators of significant new adverse effects or risks (§ 201.327(i)(1)(i)) and new safety and other observations (§ 201.327(i)(1)(v)) 20 40 800 0.5 (30 minutes) 400 Investigators provide safety reports in accordance with § 201.327(i)(1)(vii)(A) 20 24.4 488 0.5 (30 minutes) 244 Investigators provide a final report in accordance with § 201.327(i)(1)(vii)(B) (one time per study) 20 48.75 975 3 2,925 Total 3,569 1 There are no capital costs or operating and maintenance costs associated with this collection of information. Table 10—Estimated Annual Recordkeeping Burden 1
Activity and 21 CFR section Number of recordkeepers Number of records per recordkeeper Total annual records Average burden per recordkeeping Total hours Obtain and keep a signed investigator statement and CV in accordance with § 201.327(i)(1)(i) and (iv)(B) 20 2 40 0.6 (36 minutes) 24 Generate SPF testing records for existing products (§ 201.327(l)(2)) (one-time) 20 85.5 1,710 24 41,040 Generate SPF testing records for new formulations (§ 201.327(l)(2)) 20 48.75 975 24 23,400 Keep SPF testing records for existing products (§ 201.327(l)(2)) (one-time) 782 2.1867 1,710 0.33 (20 minutes) 564.3 Keep SPF testing records for new formulations (§ 201.327(l)(2)) 782 1.2468 975 0.33 (20 minutes) 321.75 Generate Broad Spectrum testing records for existing products (§ 201.327(l)(3)) 20 203.9 4,078 1.5 6,117 Generate Broad Spectrum testing records for new formulations (§ 201.327(l)(3) 20 48.75 975 1.5 1,462.5 Keep Broad Spectrum testing records for existing products (§ 201.327(l)(3)) 782 5.215 4,078 0.17 (10 minutes) 693.3 Keep Broad Spectrum testing records for new formulations (§ 201.327(l)(3)) 782 1.2468 975 0.17 (10 minutes) 165.75 Total 773,788.6 1 There are no capital costs or operating or maintenance costs associated with this collection of information. Table 11—Estimated Annual Third-Party Disclosure Burden 1
Activity and 21 CFR section Number of respondents Number of disclosures per respondent Total annual disclosures Average burden per disclosure Total hours Create PDP labeling in accordance with statement of identity and SPF value requirements (§ 201.327(b)(1), (b)(2) and (h)(1)) for currently marketed sunscreen formulations (one-time burden) 772 5.2824 4,078 0.5 (30 minutes) 2,039 Create PDP labeling in accordance with statement of identity and SPF value requirements (§ 201.327(b)(1), (b)(2), and (h)(1)) for new formulations 772 1.943 1,500 0.5 (30 minutes) 750 Conduct SPF testing in accordance with § 201.327(i) to determine SPF value for currently marketed sunscreen formulations (if not already done) (one-time burden) 20 111 2,220 24 53,280 Conduct SPF testing in accordance with § 201.327(i) to determine SPF value for new sunscreen formulations 20 48.75 975 24 23,400 Total 79,469 1 There are no capital costs or operating or maintenance costs associated with this collection of information. In compliance with the PRA (44 U.S.C. 3407(d)), the Agency has submitted the information collection provisions of this proposed rule to OMB for review. These requirements will not be effective until FDA obtains OMB approval. FDA will publish a notice concerning OMB approval of these requirements in the Federal Register.
XVI. Federalism
We have analyzed this proposed rule in accordance with the principles set forth in Executive Order 13132. Section 4(a) of the Executive order requires Agencies to “construe . . . a Federal statute to preempt State law only where the statute contains an express preemption provision or there is some other clear evidence that the Congress intended preemption of State law, or where the exercise of State authority conflicts with the exercise of Federal authority under the Federal statute.” The sole statutory provision giving preemptive effect to this proposed rule is section 751 of the FD&C Act (21 U.S.C. 379r). We have complied with all of the applicable requirements under the Executive order and have determined that the preemptive effect of this proposed rule, if finalized, would be consistent with Executive Order 13132. Through publication of this proposed rule, we are providing notice and an opportunity for State and local officials to comment on this rulemaking.
XVII. Consultation and Coordination With Indian Tribal Governments
We have analyzed this proposed rule in accordance with the principles set forth in Executive Order 13175. We have tentatively determined that the rule does not contain policies that would have a substantial direct effect on one or more Indian Tribes, on the relationship between the Federal Government and Indian Tribes, or on the distribution of power and responsibilities between the Federal Start Printed Page 6258Government and Indian Tribes. The Agency solicits comments from tribal officials on any potential impact on Indian Tribes from this proposed action.
XVIII. References
The following references marked with an asterisk (*) are on display at the Dockets Management Staff (see ADDRESSES) and are available for viewing by interested persons between 9 a.m. and 4 p.m., Monday through Friday; they also are available electronically at https://www.regulations.gov. References without asterisks are not on public display at https://www.regulations.gov because they have copyright restriction. Some may be available at the website address, if listed. References without asterisks are available for viewing only at the Dockets Management Staff. FDA has verified the website addresses, as of the date this document publishes in the Federal Register, but websites are subject to change over time.
* 1. FDA, Guidance for Industry, “Enforcement Policy—OTC Sunscreen Drug Products Marketed Without an Approved Application,” May 2018 (available at https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM259001), accessed June 7, 2018.
2. Matlack, S., “From Tanning Accessory to Health Necessity: History of the OTC Sunscreen Monograph in Light of the Sunscreen Revolution” (available at https://nrs.harvard.edu/urn-3:HUL.InstRepos:8965570), accessed March 27, 2018.
* 3. National Cancer Institute, “Cancer Trends Progress Report,” February 2018 (available at https://progressreport.cancer.gov), accessed July 27, 2018.
4. Urbach, F., “The Historical Aspects of Sunscreens,” Journal of Photochemistry and Photobiology B, vol. 64(2-3), pp. 99-104, 2001.
* 5. U.S. Department of Health and Human Services, “The Surgeon General's Call to Action to Prevent Skin Cancer,” (available at https://www.surgeongeneral.gov/library/calls/prevent-skin-cancer/exec-summary.html), accessed April 20, 2018.
6. Svarc, F., “A Brief Illustrated History on Sunscreens and Sun Protection,” Pure and Applied Chemistry, vol. 87(9-10), pp. 929-936, 2015.
* 7. FDA, “Sunscreen: How to Help Protect Your Skin from the Sun” (available at https://www.fda.gov/drugs/resourcesforyou/consumers/buyingusingmedicinesafely/understandingover-the-countermedicines/ucm239463.htm), accessed March 27, 2018.
* 8. Centers for Disease Control and Prevention (CDC), “Skin Cancer: Sun Safety” (available at https://www.cdc.gov/cancer/skin/basic_info/sun-safety.htm), accessed March 27, 2018.
9. American Cancer Society, “Melanoma Skin Cancer” (available at https://www.cancer.org/cancer/melanoma-skin-cancer.html?gclid=CMCkj93Uqs4CFYtahgodxIwIYA&mkwid=s_dc;; https://www.cancer.org/cancer/melanoma-skin-cancer.html), accessed March 27, 2018.
* 10. FDA, “Nonprescription Drug Advisory Committee, September 4-5, 2014, Meeting Minutes” (available at https://wayback.archive-it.org/7993/20170404152726/https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/NonprescriptionDrugsAdvisoryCommittee/UCM421304.pdf), accessed March 27, 2018.
* 11. FDA, Guidance for Industry, “Nonprescription Sunscreen Drug Products—Safety and Effectiveness Data,” November 2016 (available at https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM473464.pdf), accessed March 27, 2018.
* 12. FDA, Guidance for Industry, “ICH S10 Photosafety Evaluation of Pharmaceuticals,” January 2015 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm337572.pdf), accessed March 27, 2018.
13. Bashaw, E.D., D.C. Tran, C.G. Shukla, et al., “Maximal Usage Trial: An Overview of the Design of Systemic Bioavailability Trial for Topical Dermatological Products,” Therapeutic Innovation and Regulatory Science, vol. 49(1), pp. 108-115, 2015.
14. Bassani, A.S., D. Banov, and H. Phan, “Characterization of the Percutaneous Absorption of Ketoprofen Using the Franz Skin Finite Dose Model,” Postgraduate Medical Journal, vol. 128(2), pp. 262-267, 2017.
15. U.S. Pharmacopeia, “1724: Semisolid Drug Products—Performance Tests,” US Pharmacopeia 40—National Formulary 35, pp. 2055-2067, December 2017.
* 16. FDA, Draft Guidance for Industry, “Maximal Usage Trials for Topical Active Ingredients Being Considered for Inclusion in an Over-The-Counter Monograph: Study Elements and Considerations,” May 2018 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm608356.pdf), accessed June 7, 2018.
* 17. FDA, Guidance for Industry, “The Need for Long-term Rodent Carcinogenicity Studies of Pharmaceuticals,” March 1996 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm074911.pdf), accessed March 27, 2018.
18. Jacobs, A.C. and P.C. Brown, “Regulatory Forum Opinion Piece; Transgenic/Alternative Carcinogenicity Assays: A Retrospective Review of Studies Submitted to CDER/FDA 1997-2014,” Toxicology Pathology, vol. 43(5), pp. 605-610, 2015.
* 19. FDA, Guidance for Industry, “M7(R1) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk,” March 2018 (available at https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM347725.pdf), accessed March 27, 2018.
* 20. FDA, Guidance for Industry, “ICH S5A Detection of Toxicity to Reproduction for Medicinal Products,” September 1994 (available at https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074950.pdf), accessed March 27, 2018.
* 21. FDA, Guidance for Industry, “Nonclinical Evaluation of Endocrine-Related Drug Toxicity,” September 2015 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm369043.pdf), accessed March 27, 2018.
* 22. FDA, Guidance for Industry, “Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies,” March 1995 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm074937.pdf), accessed March 27, 2018.
* 23. National Science and Technology Council Committee on Technology, “The National Nanotechnology Initiative Strategic Plan,” February 2014 (available at http://www.nano.gov/sites/default/files/pub_resource/2014_nni_strategic_plan.pdf), accessed March 27, 2018.
* 24. FDA, Guidance for Industry, “Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology,” June 2014 (available at http://www.fda.gov/RegulatoryInformation/Guidances/ucm257698.htm), accessed March 27, 2018.
25. Hayden, D.R., A. Imhof, and K.P. Velikov, “Biobased Nanoparticles for Broadband UV Protection with Photostabilized UV Filters,” American Chemical Society Applied Materials and Interfaces, vol. 8(48), pp. 32655-32660, 2016.
26. Beasley, D.G. and T.A. Meyer, “Characterization of the UVA Protection Provided by Avobenzone, Zinc Oxide, and Titanium Dioxide in Broad-Spectrum Sunscreen Products,” American Journal of Clinical Dermatology, vol. 11(6), pp. 413-421, 2010.
27. Fairhurst, D. and M.A. Mitchnick, “Particulate Sun Blocks: General Principles,” N.J. Lowe, N.A. Shaath, M.A. Pathak, editors, Sunscreens: Development, Evaluation, and Regulatory Aspects, 2nd ed., New York: M. Dekker, pp. 313-352, 1997.
28. Mitchnick, M.A., D. Fairhurst, and S.R. Pinnell, “Microfine Zinc Oxide (Z-Cote) as a Photostable UVA/UVB Sunblock Start Printed Page 6259Agent,” Journal of the American Academy of Dermatology, vol. 40(1), pp. 85-90, 1999.
29. Agren, M.S., “Studies on Zinc in Wound Healing,” Acta Dermato-Venereologica. Supplementum, vol. 154, pp. 1-36, 1990.
30. Samman, S. “Zinc.” J.I. Mann and A.S. Truswell, editors, Essentials of Human Nutrition, 3rd ed., Oxford: Oxford University Press, pp. 138-142, 2007.
31. Plum, L.M., L. Rink, and H. Haase, “The Essential Toxin: Impact of Zinc on Human Health,” International Journal of Environmental Research and Public Health, vol. 7(4), pp. 1342-1365, 2010.
32. Leite-Silva, V.R., M. Le Lamer, W.Y. Sanchez, et al., “The Effect of Formulation on the Penetration of Coated and Uncoated Zinc Oxide Nanoparticles into the Viable Epidermis of Human Skin in Vivo,” European Journal of Pharmaceutics and Biopharmaceutics, vol. 84(2), pp. 297-308, 2013.
33. Darvin, M.E., K. Konig, M. Kellner-Hoefer, et al., “Safety Assessment by Multiphoton Fluorescence/Second Harmonic Generation/Hyper-Rayleigh Scattering Tomography of ZnO Nanoparticles Used in Cosmetic Products,” Skin Pharmacology and Physiology, vol. 25(4), pp. 219-226, 2012.
34. Gulson, B., M. McCall, M. Korsch, et al., “Small amounts of zinc from zinc oxide particles in sunscreens applied outdoors are absorbed through human skin,” Toxicological Sciences, vol. 118(1), pp. 140-149, 2010.
35. Gulson, B., H. Wong, M. Korsch, et al., “Comparison of dermal absorption of zinc from different sunscreen formulations and differing UV exposure based on stable isotope tracing,” The Science of the Total Environment, vol. 420, pp. 313-318, 2012.
36. Stefanidou, M., C. Maravelias, A. Dona, et al., “Zinc: A Multipurpose Trace Element,” Archives of Toxicology, vol. 80(1), pp. 1-9, 2006.
37. Monteiro-Riviere, N.A., K. Wiench, R. Landsiedel, et al., “Safety Evaluation of Sunscreen Formulations Containing Titanium Dioxide and Zinc Oxide Nanoparticles in UVB Sunburned Skin: An in Vitro and in Vivo Study,” Toxicological Sciences, vol. 123(1), pp. 264-280, 2011.
38. Agren, M.S., “Percutaneous Absorption of Zinc from Zinc Oxide Applied Topically to Intact Skin in Man,” Dermatologica, vol. 180(1), pp. 36-39, 1990.
39. Agren, M.S., “Influence of Two Vehicles for Zinc Oxide on Zinc Absorption through Intact Skin and Wounds,” Acta Dermato-Venereologica, vol. 71(2), pp. 153-156, 1991.
* 40. European Commission, SCCS, “Opinion on Zinc Oxide (nano form)” (available at http://ec.europa.eu/health/scientific_committees/consumer_safety/docs/sccs_o_103.pdf), accessed April 4, 2018.
41. Beeckman, D., L. Schoonhoven, S. Verhaeghe, et al., “Prevention and Treatment of Incontinence-Associated Dermatitis: Literature Review,” Journal of Advanced Nursing, vol. 65(6), pp. 1141-1154, 2009.
42. Li, C.H., C.C. Shen, Y.W. Cheng, et al., “Organ Biodistribution, Clearance, and Genotoxicity of Orally Administered Zinc Oxide Nanoparticles in Mice,” Nanotoxicology, vol. 6(7), pp. 746-756, 2012.
43. Ryu, H.J., M.Y. Seo, S.K. Jung, et al., “Zinc Oxide Nanoparticles: A 90-Day Repeated-Dose Dermal Toxicity Study in Rats,” International Journal of Nanomedicine, vol. 9 Suppl 2, pp. 137-144, 2014.
44. Hong, J.S., M.K. Park, M.S. Kim, et al., “Effect of Zinc Oxide Nanoparticles on Dams and Embryo-Fetal Development in Rats,” International Journal of Nanomedicine, vol. 9 Suppl 2, pp. 145-157, 2014;
45. Hong, J.S., M.K. Park, M.S. Kim, et al., “Prenatal Development Toxicity Study of Zinc Oxide Nanoparticles in Rats,” International Journal of Nanomedicine, vol. 9 Suppl 2, pp. 159-171, 2014.
* 46. FDA, “Inactive Ingredient Search for Approved Drug Products” (available at https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm), accessed March 27, 2018.
47. Gad, S.C., “Titanium.” P. Wexler, editors, Encyclopedia of Toxicology (Third Edition), Oxford: Academic Press, 2014: 584-585.
48. Pflucker, F., V. Wendel, H. Hohenberg, et al., “The Human Stratum Corneum Layer: An Effective Barrier against Dermal Uptake of Different Forms of Topically Applied Micronised Titanium Dioxide,” Skin Pharmacology and Applied Skin Physiology, vol. 14 Suppl 1, pp. 92-97, 2001.
49. Schulz, J., H. Hohenberg, F. Pflucker, et al., “Distribution of Sunscreens on Skin,” Advanced Drug Delivery Reviews, vol. 54 Suppl 1, pp. S157-163, 2002.
50. Sandstead, H.H., “Zinc.” B.A. Fowler and M. Nordberg, editors, Handbook on the Toxicology of Metals (Fourth Edition), San Diego: Academic Press, 2015: 1369-1385.
51. Gotman, I., “Characteristics of Metals Used in Implants,” Journal of Endourology, vol. 11(6), pp. 383-389, 2009.
52. Ipach, I., R. Schafer, F. Mittag, et al., “The Development of Whole Blood Titanium Levels after Instrumented Spinal Fusion—Is There a Correlation between the Number of Fused Segments and Titanium Levels?,” BioMed Central Musculoskeletal Disorders, vol. 13, pp. 159, 2012.
53. Sadrieh, N., A.M. Wokovich, N.V. Gopee, et al., “Lack of Significant Dermal Penetration of Titanium Dioxide from Sunscreen Formulations Containing Nano- and Submicron-Size Tio2 Particles,” Toxicological Sciences, vol. 115(1), pp. 156-166, 2010.
54. Kiss, B., T. Biro, G. Czifra, et al., “Investigation of Micronized Titanium Dioxide Penetration in Human Skin Xenografts and Its Effect on Cellular Functions of Human Skin-Derived Cells,” Experimental Dermatology, vol. 17(8), pp. 659-667, 2008.
55. Crosera, M., A. Prodi, M. Mauro, et al., “Titanium Dioxide Nanoparticle Penetration into the Skin and Effects on HaCaT Cells,” International Journal of Environmental Research and Public Health, vol. 12(8), pp. 9282-9297, 2015.
56. Warheit, D.B., R. Boatman, and S.C. Brown, “Developmental Toxicity Studies with 6 Forms of Titanium Dioxide Test Materials (3 Pigment-Different Grade & 3 Nanoscale) Demonstrate an Absence of Effects in Orally-Exposed Rats,” Regulatory Toxicology and Pharmacology, vol. 73(3), pp. 887-896, 2015.
57. Adachi, K., N. Yamada, K. Yamamoto, et al., “In Vivo Effect of Industrial Titanium Dioxide Nanoparticles Experimentally Exposed to Hairless Rat Skin,” Nanotoxicology, vol. 4(3), pp. 296-306, 2010.
58. Adachi, K., N. Yamada, Y. Yoshida, et al., “Subchronic Exposure of Titanium Dioxide Nanoparticles to Hairless Rat Skin,” Experimental Dermatology, vol. 22(4), pp. 278-283, 2013.
59. Unnithan, J., M.U. Rehman, F.J. Ahmad, et al., “Aqueous Synthesis and Concentration-Dependent Dermal Toxicity of Tio2 Nanoparticles in Wistar Rats,” Biological Trace Element Research, vol. 143(3), pp. 1682-1694, 2011.
60. Unnithan, J., M.U. Rehman, F.J. Ahmad, et al., “Concentration Dependent Toxicity of Approximately 20 Nm Anatase Titanium Dioxide Nanoparticles—an in Vivo Study on Wistar Rats,” Journal of Biomedical Nanotechnology, vol. 7(1), pp. 207-208, 2011.
61. Fujishima, A., T.N. Rao, and D.A. Tryk, “Titanium dioxide photocatalysis,” Journal of Photochemistry and Photobiology C: Photochemistry Reviews, vol. 1(1), pp. 1-21, 2000.
* 62. European Commission, Scientific Committee on Consumer Safety (SCCS), “Opinion on Titanium Dioxide (nano form),” April 22, 2014, revision (available at http://ec.europa.eu/health/scientific_committees/consumer_safety/docs/sccs_o_136.pdf), accessed March 27, 2018.
* 63. FDA, Preliminary Regulatory Impact Analysis, “Sunscreen Drug Products for Over-the-Counter Human Use” (available at https://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/EconomicAnalyses/default.htm.
64. Balogh, T.S., M.V. Velasco, C.A. Pedriali, et al., “Ultraviolet Radiation Protection: Current Available Resources in Photoprotection,” Anais Brasileiros de Dermatologia, vol. 86(4), pp. 732-742, 2011.
65. Gao, L., Y. Hu, C. Ni, et al., “Retrospective Study of Photopatch Testing in a Chinese Population During a 7-Year Period,” Dermatitis, vol. 25(1), pp. 22-26, 2014.
66. Trevisi, P., C. Vincenzi, C. Chieregato, et al., “Sunscreen Sensitization: A Three-Year Study,” Dermatology (Basel, Switzerland), vol. 189(1), pp. 55-57, 1994.Start Printed Page 6260
67. Victor, F.C., D.E. Cohen, and N.A. Soter, “A 20-Year Analysis of Previous and Emerging Allergens That Elicit Photoallergic Contact Dermatitis,” Journal of the American Academy of Dermatology, vol. 62(4), pp. 605-610, 2010.
68. Teplitz, R.W., A.M. Glazer, R. M. Svoboda, et al., “Trends in US Sunscreen Formulations: Impact of Increasing Spray Usage,” Journal of the American Academy of Dermatology, vol. 78(1), pp. 187-189, 2018.
69. Willis, I., “Sensitization Potential of Para-Aminobenzoic Acid,” Cosmetics and Toiletries, vol. 91(3), pp. 63-64, 1976.
70. Fisher, A.A., “Sunscreen Dermatitis: Para-Aminobenzoic Acid and its Derivatives,” Cutis, vol. 50(3), pp. 190-192, 1992.
71. Mackie, B.S., and L.E. Mackie, “The PABA Story,” The Australasian Journal of Dermatology, vol. 40(1), pp. 51-53, 1999.
72. Pathak, M.A., “Sunscreens: Topical and Systemic Approaches for Protection of Human Skin against Harmful Effects of Solar Radiation,” Journal of the American Academy of Dermatology, vol. 7(3), pp. 285-312, 1982.
73. Wang, L.H., W.S. Huang, and H.M. Tai, “Simultaneous Determination of P-Aminobenzoic Acid and its Metabolites in the Urine of Volunteers, Treated with P-Aminobenzoic Acid Sunscreen Formulation,” Journal of Pharmaceutical and Biomedical Analysis, vol. 43(4), pp. 1430-1436, 2007.
74. Borum, M., E. Nsien, and H. Zimmerman, “Hepatotoxicity from Paraaminobenzoic Acid,” Digestive Diseases and Sciences, vol. 36(12), pp. 1793, 1991.
75. Alexopoulos, E., E. Papagianni, M. Leontsini, et al., “Para-Amino-Benzoic Acid (PABA) and Chronic Tubulointerstitial Nephritis,” Clinical Nephrology, vol. 40(6), pp. 359-360, 1993.
76. Rose, F.A. and D.R. Wiemer, “Platelet Coagulopathy Secondary to Topical Salicylate Use,” Annals of Plastic Surgery, vol. 11(4), pp. 340-343, 1983.
77. Madan, R.K. and J. Levitt, “A Review of Toxicity from Topical Salicylic Acid Preparations,” Journal of the American Academy of Dermatology, vol. 70(4), pp. 788-792, 2014.
78. Littleton, F., Jr., “Warfarin and Topical Salicylates,” Journal of the American Medical Society, vol. 263(21), p. 2888, 1990.
* 79. National Toxicology Program, NTP Technical Report 518, “Toxicology and Carcinogenesis Studies of Triethanolamine in B6C3F1 Mice (Dermal Study),” May 2004, (available at https://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/tr518.pdf), accessed March 27, 2018.
* 80. National Toxicology Program, Technical Report 449, “Toxicology and Carcinogenesis Studies of Triethanolamine in F344/N Rats and B6C3F1 Mice (Dermal Studies),” November 1999, (available at https://ntp.niehs.nih.gov/ntp/htdocs/lt_rpts/tr449.pdf), accessed March 27, 2018.
81. Benson, H.A., V. Sarveiya, S. Risk, et al., “Influence of Anatomical Site and Topical Formulation on Skin Penetration of Sunscreens,” Therapeutics and Clinical Risk Management, vol. 1(3), pp. 209-218, 2005.
82. Janjua, N.R., B. Kongshoj, A.M. Andersson, et al., “Sunscreens in Human Plasma and Urine after Repeated Whole-Body Topical Application,” Journal of the European Academy of Dermatology and Venereology, vol. 22(4), pp. 456-461, 2008.
83. Janjua, N.R., B. Mogensen, A.M. Andersson, et al., “Systemic Absorption of the Sunscreens Benzophenone-3, Octyl-Methoxycinnamate, and 3-(4-Methyl-Benzylidene) Camphor after Whole-Body Topical Application and Reproductive Hormone Levels in Humans,” The Journal of Investigative Dermatology, vol. 123(1), pp. 57-61, 2004.
84. Sarveiya, V., S. Risk, and H. Benson, “Liquid Chromatographic Assay for Common Sunscreen Agents: Application to In Vivo Assessment of Skin Penetration and Systemic Absorption in Human Volunteers,” Journal of Chromatography, vol. 803(2), pp. 225-231, 2004.
85. Pastore, M.N., Y.N. Kalia, M. Horstmann, et al., “Transdermal Patches: History, Development and Pharmacology,” British Journal of Pharmacology, vol. 172(9), pp. 2179-2209, 2015.
86. Gomez, E., A. Pillon, H. Fenet, et al., “Estrogenic Activity of Cosmetic Components in Reporter Cell Lines: Parabens, UV Screens, and Musks,” Journal of Toxicology and Environmental Health, vol. 68(4), pp. 239-251, 2005.
87. Schreurs, R., P. Lanser, W. Seinen, et al., “Estrogenic Activity of UV Filters Determined by an in Vitro Reporter Gene Assay and an in Vivo Transgenic Zebrafish Assay,” Archives of Toxicology, vol. 76(5-6), pp. 257-261, 2002
88. Schlumpf, M., B. Cotton, M. Conscience, et al., “In Vitro and in Vivo Estrogenicity of UV Screens,” Environmental Health Perspectives, vol. 109(3), pp. 239-244, 2001
* 89. European Commission, SCCNFP, “Opinion on the Evaluation of Potentially Estrogenic Effects of UV-Filters Adopted by the SCCNFP During the 17th Plenary Meeting of 12 June 2001” (available at (http://ec.europa.eu/health/scientific_committees/consumer_safety/opinions/sccnfp_opinions_97_04/sccp_out145_en.htm), accessed March 27, 2018.
90. Schreurs, R.H., E. Sonneveld, J.H. Jansen, et al., “Interaction of Polycyclic Musks and UV Filters with the Estrogen Receptor (Er), Androgen Receptor (Ar), and Progesterone Receptor (Pr) in Reporter Gene Bioassays,” Toxicological Sciences, vol. 83(2), pp. 264-272, 2005.
91. Ma, R., B. Cotton, W. Lichtensteiger, et al., “UV Filters with Antagonistic Action at Androgen Receptors in the MDA-kb2 Cell Transcriptional-Activation Assay,” Toxicological Sciences, vol. 74(1), pp. 43-50, 2003.
92. Krause, M., A. Klit, M. Blomberg Jensen, et al., “Sunscreens: Are They Beneficial for Health? An Overview of Endocrine Disrupting Properties of UV-Filters,” International Journal of Andrology, vol. 35(3), pp. 424-436, 2012
93. Szwarcfarb, B., S. Carbone, R. Reynoso, et al., “Octyl-Methoxycinnamate (OMC), an Ultraviolet (UV) Filter, Alters LHRH and Amino Acid Neurotransmitters Release from Hypothalamus of Immature Rats,” Experimental and Clinical Endocrinology and Diabetes, vol. 116(2), pp. 94-98, 2008
94. Seidlova-Wuttke, D., H. Jarry, J. Christoffel, et al., “Comparison of Effects of Estradiol (E2) with Those of Octylmethoxycinnamate (OMC) and 4-Methylbenzylidene Camphor (4MBC)—2 Filters of UV Light—on Several Uterine, Vaginal and Bone Parameters,” Toxicology and Applied Pharmacology, vol. 210(3), pp. 246-254, 2006.
95. Freitas, J.V., F.S. Praca, M.V. Bentley, et al., “Trans-Resveratrol and Beta-Carotene from Sunscreens Penetrate Viable Skin Layers and Reduce Cutaneous Penetration of UV-Filters,” International Journal of Pharmaceutics, vol. 48(1-2), pp. 131-137, 2015.
96. Montenegro, L. and G. Puglisi, “Evaluation of Sunscreen Safety by in Vitro Skin Permeation Studies: Effects of Vehicle Composition,” Die Pharmazie, vol. 68(1), pp. 34-40, 2013.
97. Golmohammadzadeh, S., M.R. Jaafarixx, and N. Khalili, “Evaluation of Liposomal and Conventional Formulations of Octyl Methoxycinnamate on Human Percutaneous Absorption Using the Stripping Method,” Journal of Cosmetic Science, vol. 59(5), pp. 385-398, 2008.
98. Montenegro, L., C. Carbone, D. Paolino, et al., “In Vitro Skin Permeation of Sunscreen Agents from O/W Emulsions,” International Journal of Cosmetic Science, vol. 30(1), pp. 57-65, 2008.
99. Gupta, V.K., J.L. Zatz, and M. Rerek, “Percutaneous Absorption of Sunscreens through Micro-Yucatan Pig Skin in Vitro,” Pharmaceutical Research, vol. 16(10), pp. 1602-1607, 1999.
100. Hayden, C.G., S.E. Cross, C. Anderson, et al., “Sunscreen Penetration of Human Skin and Related Keratinocyte Toxicity after Topical Application,” Skin Pharmacology and Physiology, vol. 18(4), pp. 170-174, 2005.
101. Treffel, P. and B. Gabard, “Skin Penetration and Sun Protection Factor of Ultra-Violet Filters from Two Vehicles,” Pharmaceutical Research, vol. 13(5), pp. 770-774, 1996.
102. Jiang, R., M.S. Roberts, R.J. Prankerd, et al., “Percutaneous Absorption of Sunscreen Agents from Liquid Paraffin: Self-Association of Octyl Salicylate and Effects on Skin Flux,” Journal of Pharmaceutical Sciences, vol. 86(7), pp. 791-796, 1997.
103. Walters, K.A., A.C. Watkinson, and K.R. Brain, “In Vitro Skin Permeation Evaluation: The Only Realistic Option,” International Journal of Cosmetic Science, vol. 20(5), pp. 307-316, 1998.Start Printed Page 6261
104. Walters, K.A., K.R. Brain, D. Howes, et al., “Percutaneous Penetration of Octyl Salicylate from Representative Sunscreen Formulations through Human Skin in Vitro,” Food and Chemical Toxicology, vol. 35(12), pp. 1219-1225, 1997.
105. Santos, P., A.C. Watkinson, J. Hadgraft, et al., “Influence of Penetration Enhancer on Drug Permeation from Volatile Formulations,” International Journal of Pharmaceuticals, vol. 439, pp. 260-268, 2012.
106. Potard, G., C. Laugel, H. Schaefer, et al., “The Stripping Technique: In Vitro Absorption and Penetration of Five UV Filters on Excised Fresh Human Skin,” Skin Pharmacology and Applied Skin Physiology, vol. 13(6), pp. 336-344, 2000.
107. Kurul, E. and S. Hekimoglu, “Skin Permeation of Two Different Benzophenone Derivatives from Various Vehicles,” International Journal of Cosmetic Science, vol. 23(4), pp. 211-218, 2001.
108. Brinon, L., S. Geiger, V. Alard, et al., “Percutaneous Absorption of Sunscreens from Liquid Crystalline Phases,” Journal of Controlled Release, vol. 60(1), pp. 67-76, 1999.
109. Burnett, M.E. and S.Q. Wang, “Current Sunscreen Controversies: A Critical Review,” Photodermatology, Photoimmunology and Photomedicine, vol. 27(2), pp. 58-67, 2011.
110. Palm, M.D. and M.N. O'Donoghue, “Update on Photoprotection,” Dermatologic Therapy, vol. 25(5), pp. 360-376, 2007.
* 111. European Commission. Scientific Committee on Consumer Products (SCCP), “Opinion on Benzophenone-3,” 2008 (available at https://ec.europa.eu/health/ph_risk/committees/04_sccp/docs/sccp_o_159.pdf), accessed March 27, 2018.
112. Environmental Working Group, “CDC: Americans Carry Body Burden of Toxic Sunscreen Chemical,” March 2008 (available at https://www.ewg.org/research/cdc-americans-carry-body-burden-toxic-sunscreen-chemical#.WrpKP8a1tPZ), accessed March 27, 2018.
* 113. National Library of Medicine, “Household Products Database,” 2007 (available at https://hpd.nlm.nih.gov/index.htm), accessed March 27, 2018.
114. Kadry, A.M., C.S. Okereke, M.S. Abdel-Rahman, et al., “Pharmacokinetics of Benzophenone-3 after Oral Exposure in Male Rats,” Journal of Applied Toxicology, vol. 15(2), pp. 97-102, 1995.
115. Calafat, A.M., L.Y. Wong, X. Ye, et al., “Concentrations of the Sunscreen Agent Benzophenone-3 in Residents of the United States: National Health and Nutrition Examination Survey 2003-2004,” Environmental Health Perspectives, vol. 116(7), pp. 893-897, 2008.
116. Gustavsson Gonzalez, H., A. Farbrot, and O. Larko, “Percutaneous Absorption of Benzophenone-3, a Common Component of Topical Sunscreens,” Clinical and Experimental Dermatology, vol. 27(8), pp. 691-694, 2002.
117. Schlumpf, M., P. Schmid, S. Durrer, et al., “Endocrine Activity and Developmental Toxicity of Cosmetic UV Filters—an Update,” Toxicology, vol. 205(1-2), pp. 113-122, 2004.
118. French, J.E., “NTP Technical Report on the Toxicity Studies of 2-Hydroxy-4-Methoxybenzophenone (Cas No. 131-57-7) Adminstered Topically and in Dosed Feed to F344/N Rats and B6c3f1 Mice,” Toxicity Report Series, vol. 21, pp. 1-e14, 1992.
119. Nakamura, N., A.L. Inselman, G.A. White, et al., “Effects of Maternal and Lactational Exposure to 2-Hydroxy-4-Methoxybenzone on Development and Reproductive Organs in Male and Female Rat Offspring,” Birth Defects Research. Part B, Developmental and Reproductive Toxicology, vol. 104(1), pp. 35-51, 2015.
120. Janjua, N.R., B. Kongshoj, J.H. Petersen, et al., “Sunscreens and Thyroid Function in Humans after Short-Term Whole-Body Topical Application: A Single-Blinded Study,” The British Journal of Dermatology, vol. 156(5), pp. 1080-1082, 2007.
121. Schiffer, C., A. Muller, D.L. Egeberg, et al., “Direct Action of Endocrine Disrupting Chemicals on Human Sperm,” European Molecular Biology Organization Reports, vol. 15(7), pp. 758-765, 2014.
* 122. National Toxicology Program, “Testing Status of 2-Hydroxy-4-methoxybenzophenone 10260-S” (available at https://ntp.niehs.nih.gov/testing/status/agents/ts-10260-s.html), accessed March 27, 2018.
* 123. National Toxicology Program, “Nomination Summary for 2-Hydroxy-4-methoxybenzophenone (N79456),” 1979 (available at https://ntp.niehs.nih.gov/testing/noms/search/summary/nm-n79456.html), accessed March 27, 2018.
124. Hanson, K.M., E. Gratton, and C.J. Bardeen, “Sunscreen Enhancement of UV-Induced Reactive Oxygen Species in the Skin,” Free Radical Biology and Medicine, vol. 41(8), pp. 1205-1212, 2006.
125. Warshaw, E.M., M.Z. Wang, H.I. Maibach, et al., “Patch Test Reactions Associated with Sunscreen Products and the Importance of Testing to an Expanded Series: Retrospective Analysis of North American Contact Dermatitis Group Data, 2001 to 2010,” Dermatitis, vol. 24(4), pp. 176-182, 2013.
126. Ang, P., S.K. Ng, and C.L. Goh, “Sunscreen Allergy in Singapore,” American Journal of Contact Dermatitis, vol. 9(1), pp. 42-44, 1998.
127. Bruynzeel, D.P., J. Ferguson, K. Andersen, et al., “Photopatch Testing: A Consensus Methodology for Europe,” Journal of the European Academy of Dermatology and Venereology, vol. 18(6), pp. 679-682, 2004.
128. Bryden, A.M., H. Moseley, S.H. Ibbotson, et al., “Photopatch Testing of 1155 Patients: Results of the U.K. Multicentre Photopatch Study Group,” British Journal of Dermatology, vol. 155(4), pp. 737-747, 2006.
129. Dromgoole, S.H. and H.I. Maibach, “Sunscreening Agent Intolerance: Contact and Photocontact Sensitization and Contact Urticaria,” Journal of the American Academy of Dermatology, vol. 22(6 Pt 1), pp. 1068-1078, 1990.
130. European Multicentre Photopatch Test Study (EMCPPTS) Taskforce, “A European Multicentre Photopatch Test Study,” The British Journal of Dermatology, vol. 166(5), pp. 1002-1009, 2012.
131. Heurung, A.R., S.I. Raju, and E. M. Warshaw, “Benzophenones,” Dermatitis, vol. 25(1), pp. 3-10, 2014.
132. Heurung, A.R., S.I. Raju, and E.M. Warshaw, “Adverse Reactions to Sunscreen Agents: Epidemiology, Responsible Irritants and Allergens, Clinical Characteristics, and Management,” Dermatitis, vol. 25(6), pp. 289-326, 2014.
133. Journe, F., M.C. Marguery, J. Rakotondrazafy, et al., “Sunscreen Sensitization: A 5-Year Study,” Acta Dermato-Venereologica, vol. 79(3), pp. 211-213, 1999.
134. Rodriguez, E., M.C. Valbuena, M. Rey, et al., “Causal Agents of Photoallergic Contact Dermatitis Diagnosed in the National Institute of Dermatology of Colombia,” Photodermatology, Photoimmunology and Photomedicine, vol. 22(4), pp. 189-192, 2006.
135. Schauder, S. and H. Ippen, “Contact and Photocontact Sensitivity to Sunscreens. Review of a 15-Year Experience and of the Literature,” Contact Dermatitis, vol. 37(5), pp. 221-232, 1997.
136. Valbuena Mesa, M.C. and E.V. Hoyos Jimenez, “Photopatch Testing in Bogota (Colombia): 2011-2013,” Contact Dermatitis, vol. 74(1), pp. 11-17, 2016.
137. Swedish Research Council, “Sunscreens with Benzophenone-3 Unsuitable for Children,” Public release date: November 6, 2006 (available at https://www.eurekalert.org/pub_releases/2006-11/src-swb110606.php), accessed March 27, 2018.
138. Ceresole, R., Y.K. Han, M.A. Rosasco, et al., “Drug-Excipient Compatibility Studies in Binary Mixtures of Avobenzone,” Journal of Cosmetic Science, vol. 64(5), pp. 317-328, 2013.
139. Nash, J.F., and P.R. Tanner, “Relevance of UV Filter/Sunscreen Product Photostability to Human Safety,” Photodermatology, Photoimmunology and Photomedicine, vol. 30(2-3), pp. 88-95, 2014.
140. Kockler, J., S. Robertson, M. Oelgemoller, et al., “Chapter 3: Butyl Methoxy Dibenzoylmethane.” H. G. Brittian, editor. Profiles of Drug Substances, Excipients, and Related Methodology, vol. 38, Elsevier, pp. 87-111, 2013.
141. Simeoni, S., S. Scalia, and H.A. Benson, “Influence of Cyclodextrins on in Vitro Human Skin Absorption of the Sunscreen, Butyl-Methoxydibenzoylmethane,” International Journal of Pharmaceutics, vol. 280(1-2), pp. 163-171, 2004.Start Printed Page 6262
142. Stenberg, C. and O. Larko, “Sunscreen Application and Its Importance for the Sun Protection Factor,” Archives of Dermatology, vol. 121(11), pp. 1400-1402, 1985.
143. Brown, S. and B.L. Diffey, “The Effect of Applied Thickness on Sunscreen Protection: In Vitro and In Vivo Studies,” Photochemistry and Photobiology, vol. 44(4), pp. 509-13, 1986.
144. Raney, S., P. Lehman, and T. Franz, “30th Anniversary of the Franz Cell Finite Dose Model: The Crystal Ball of Topical Drug Development,” Journal of Drug Delivery Science and Technology, vol. 8(7), pp. 32-37, 2008.
145. Ng, S.F., J.J. Rouse, F.D. Sanderson, et al., “Validation of a Static Franz Diffusion Cell System for In Vitro Permeation Studies,” AAPS PharmSciTech, vol. 11(3), pp. 1432-1441, 2010.
146. Franz, T.J., P.A. Lehman, and S.G. Raney, “Use of Excised Human Skin to Assess the Bioequivalence of Topical Products,” Skin Pharmacology and Physiology, vol. 22, pp. 276-286, 2009.
147. Nallagundla, S., S. Patnala, and I. Kanfer, “Comparison of In Vitro Release Rates of Acyclovir from Cream Formulations Using Vertical Diffusion Cells,” AAPS PharmSciTech, vol. 15(4), pp. 994-999, 2014.
148. Stuart, B.O., “Deposition and Clearance of Inhaled Particles,” Environmental Health Perspectives, vol. 55, pp. 369-390, 1984.
149. Leikauf, G.D., “Chapter 15: Toxic Responses of the Respiratory System,” C.D. Klaassen, editor. Casarett and Doull's Toxicology: The Basic Science of Poisons, 8th ed., New York, NY: McGraw-Hill; pp. 691-732, 2013.
150. Rothe, H., R. Fautz, E. Gerber, et al., “Special Aspects of Cosmetic Spray Safety Evaluations: Principles on Inhalation Risk Assessment,” Toxicology Letters, vol. 205(2), pp. 97-104, 2011.
151. Steiling W., M. Bascompta, P. Carthew, et al., “Principle Considerations for the Risk Assessment of Sprayed Consumer Products,” Toxicology Letters, vol. 227(1), pp. 41-49, 2014.
152. Brown, J.S., T. Gordon, O. Price, et al., “Thoracic and Respirable Particle Definitions for Human Health Risk Assessment,” Particle and Fibre Toxicology, vol. 10, p. 12, 2013.
153. Liu, X., D. Rua, A. Wokovich, et al., “Particle Size Distribution Analysis of OTC Drug Products with Unintended Inhalation Exposure to Consumers [abstract].” In: American Association of Pharmaceutical Scientists Annual Meeting and Exposition; November 12-15, 2017; San Diego Convention Center. Abstract number T1109, 2017.
154. Liu, X., D. Rua, A. Wokovich, et al., “Particle Size Distribution Analysis of OTC Aerosol or Powder Drug Products with Potential for Inadvertent Inhalation Exposure to Consumers,” Journal of Pharmaceutical Science, doi: 10.1016/j.xphs.2018.10.066 (Epub ahead of print), 2018, (available at https://www.ncbi.nlm.nih.gov/pubmed/30468827,, accessed December 6, 2018).
155. U.S. Pharmacopeia, Chapter 601—Inhalation and Nasal Drug Products: Aerosols, Sprays, and Powders—Performance Quality Tests, “Part B. Droplet/Particle Size Distribution—Nasal Aerosols, Sprays, and Powders,” US Pharmacopeia 40—National Formulary 35, pp. 478-480, December 2017.
* 156. FDA, “Use Sunscreen Spray? Avoid Open Flame,” (available at http://wayback.archive-it.org/7993/20180125094109/https:/www.fda.gov/ForConsumers/ConsumerUpdates/ucm359437.htm), accessed March 27, 2018.
157. Mouret, S., C. Baudouin, M. Charveron, et al., “Cyclobutane Pyrimidine Dimers Are Predominant DNA Lesions in Whole Human Skin Exposed to UVA Radiation,” Proceedings of the National Academies of Sciences of the United States of America, vol. 103(37), pp. 13765-13770, 2006.
158. McKinlay, A.F. and B.L. Diffey, “A Reference Action Spectrum for Ultraviolet Induced Erythema in Human Skin,” CIE-Journal, vol. 6, pp. 17-22, 1987.
159. Noonan, F.P., M.R. Zaidi, A. Wolnicka-Glubisz, et al., “Melanoma Induction by Ultraviolet A but Not Ultraviolet B Radiation Requires Melanin Pigment,” Nature Communications, vol. 3, pp. 884, 2012.
160. Ridley, A.J., J.R. Whiteside, T.J. McMillan, et al., “Cellular and Sub-Cellular Responses to UVA in Relation to Carcinogenesis,” International Journal of Radiation Biology, vol. 85(3), pp. 177-195, 2009.
161. Premi, S., S. Wallisch, C.M. Mano, et al., “Photochemistry. Chemiexcitation of Melanin Derivatives Induces DNA Photoproducts Long after UV Exposure,” Science, vol. 347(6224), pp. 842-847, 2015.
162. Brash, D.E., “UV-Induced Melanin Chemiexcitation: A New Mode of Melanoma Pathogenesis,” Toxicologic Pathology, vol. 44(4), pp. 552-554, 2016.
163. Hodis, E., I.R. Watson, G.V. Kryukov, et al., “A Landscape of Driver Mutations in Melanoma,” Cell, vol. 150(2), pp. 251-263, 2012.
164. Krauthammer, M., Y. Kong, B.H. Ha, et al., “Exome Sequencing Identifies Recurrent Somatic Rac1 Mutations in Melanoma,” Nature Genetics, vol. 44(9), pp. 1006, 2012.
165. Ziegler, A., A.S. Jonason, D.J. Leffell, et al., “Sunburn and P53 in the Onset of Skin-Cancer,” Nature, vol. 372(6508), pp. 773-776, 1994.
166. Tewari, A., M.M. Grage, G.I. Harrison, et al., “UVA1 Is Skin Deep: Molecular and Clinical Implications,” Photochemical and Photobiological Sciences, vol. 12(1), pp. 95-103, 2013.
167. Tewari, A., R.P. Sarkany, and A.R. Young, “UVA1 Induces Cyclobutane Pyrimidine Dimers but Not 6-4 Photoproducts in Human Skin in Vivo,” Journal of Investigative Dermatology, vol. 132(2), pp. 394-400, 2012.
168. DiGiovanna, J.J. and K.H. Kraemer, “Shining a Light on Xeroderma Pigmentosum,” Journal of Investigative Dermatology, vol. 132(3), pp. 785-796, 2012.
169. Damian, D.L., Y. J. Matthews, T.A. Phan, et al., “An Action Spectrum for Ultraviolet Radiation-Induced Immunosuppression in Humans,” British Journal of Dermatology, vol. 164(3), pp. 657-659, 2011.
170. Marionnet, C., C. Pierrard, C. Golebiewski, et al., “Diversity of Biological Effects Induced by Longwave UVA Rays (UVA1) in Reconstructed Skin,” PLoS One, vol. 9(8), 2014.
171. Autier, P., M. Boniol, and J.F. Dore, “Sunscreen Use and Increased Duration of Intentional Sun Exposure: Still a Burning Issue,” International Journal of Cancer, vol. 121(1), pp. 1-5, 2007.
172. Diffey, B., “The FDA Final Rule on Labeling and Effectiveness Testing of Sunscreens: Too Little, Too Late?” Journal of the American Academy of Dermatology, vol. 66(1), pp. 162-163, 2012.
173. Diffey, B., “Spectral Uniformity: A New Index of Broad Spectrum (UVA) Protection,” International Journal of Cosmetic Science, vol. 31(1), pp. 63-68, 2009.
174. Wang, S.Q., J.W. Stanfield, and U. Osterwalder, “In Vitro Assessments of UVA Protection by Popular Sunscreens Available in the United States,” Journal of the American Academy of Dermatology, vol. 59(6), pp. 934-942, 2008.
175. Diffey, B.L., “Sunscreens and UVA Protection: A Major Issue of Minor Importance,” Photochemistry and Photobiology, vol. 74(1), pp. 61-63, 2001.
176. Ulrich, C., J.S. Jurgensen, A. Degen, et al., “Prevention of Non-Melanoma Skin Cancer in Organ Transplant Patients by Regular Use of a Sunscreen: a 24 Months, Prospective, Case-Control Study,” The British Journal of Dermatology, vol. 161 Suppl 3, pp. 78-84, 2009.
177. Kuhn, A., K. Gensch, M. Haust, et al., “Photoprotective Effects of a Broad-Spectrum Sunscreen in Ultraviolet-Induced Cutaneous Lupus Erythematosus: a Randomized, Vehicle-Controlled, Double-Blind Study,” Journal of the American Academy of Dermatology, vol. 64(1), pp. 37-48, 2011.
178. Faurschou, A. and H.C. Wulf, “Synergistic Effect of Broad-Spectrum Sunscreens and Antihistamines in the Control of Idiopathic Solar Urticaria,” Archives of Dermatology, vol. 144(6), pp. 765-769, 2008.
179. Fourtanier, A., D. Moyal, and S. Seite, “Sunscreens Containing the Broad-Spectrum UVA Absorber, Mexoryl SX, Prevent the Cutaneous Detrimental Effects of UV Exposure: a Review of Clinical Study Results,” Photodermatology, Photoimmunology & Photomedicine, vol. 24(4), pp. 164-174, 2008.
* 180. Therapeutic Goods Administration, “Australian Regulatory Guidelines for Start Printed Page 6263Sunscreens (ARGS),” TGA Publication, pp. 1-43, 2016.
* 181. European Commission, “Commission Recommendation of 22 September 2006 on the Efficacy of Sunscreen Products and the Claims Made Relating Thereto (2006/647/EC),” Official Journal of the European Union, vol. 265, pp. 39-43, 2006 (available at http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006H0647&from=EN), accessed March 27, 2018.
182. Stanfield, J.W., H. Ou-Yang, T. Chen, et al., “Multi-Laboratory Validation of Very High Sun Protection Factor Values,” Photodermatology, Photoimmunology & Photomedicine, vol. 27(1), pp. 30-34, 2011.
183. Xu, S., M. Kwa, A. Agarwal, et al. “Sunscreen Product Performance and Other Determinants of Consumer Preferences,” Journal of the American Medical Association Dermatology, vol. 52(8), pp. 920-927, 2016.
184. Chao, L.X., S.L. Sheu, B.Y. Kong, et al. “Identifying Gaps in Consumer Knowledge About Sunscreen,” Journal of the American Academy of Dermatology, vol. 77(6), pp. 1172-1173, 2017.
185. Kong, B.Y., S.L. Sheu, and R.V. Kundu, “Assessment of Consumer Knowledge of New Sunscreen Labels,” JAMA Dermatology, vol.151(9), pp. 1028-1030, 2015.
186. Coelho, S.G., Y. Zhou, H.F. Bushar, et al., “Long-Lasting Pigmentation of Human Skin, A New Look at an Overlooked Response to UV,” Pigment Cell and Melanoma Research, vol. 22(2), pp. 238-241, 2009.
* 187. EPA, “Find the Repellent That is Right for You,” (available at https://www.epa.gov/insect-repellents/find-repellent-right-you), accessed March 27, 2018.
188. NPIC, “Insect Repellent Locator,” (available at http://pi.ace.orst.edu/repellents/), accessed March 27, 2018.
* 189. EPA, “Active Ingredients Eligible for Minimum Risk Pesticide Products” (Updated December 2015) (available at https://www.epa.gov/sites/production/files/2015-12/documents/minrisk-active-ingredients-tolerances-2015-12-15.pdf), accessed March 27, 2018.
* 190. EPA, “Using Insect Repellents Safely and Effectively” (available at https://www.epa.gov/insect-repellents/using-insect-repellents-safely-and-effectively), accessed March 27, 2018.
191. American Academy of Pediatrics, “Choosing an Insect Repellent for Your Child” (available at https://www.healthychildren.org/English/safety-prevention/at-play/Pages/Insect-Repellents.aspx), accessed March 27, 2018.
* 192. EPA, “Reregistration Eligibility Decision (R.E.D.) Facts, DEET,” April 1998 (available at https://www3.epa.gov/pesticides/chem_search/reg_actions/reregistration/fs_PC-080301_1-Apr-98.pdf), accessed March 27, 2018.
193. Katz, T.M., J.H. Miller, and A. Hebert, “Insect Repellents: Historical Perspectives and New Developments,” Journal of the American Academy of Dermatology, vol. 58(5), pp. 865-871, 2008.
*194. EPA, “3-[N-Butyl-N-acetyl]-Aminopropionic Acid, Ethyl Ester (113509) Technical Document” (available at https://www3.epa.gov/pesticides/chem_search/reg_actions/registration/related_PC-113509_1-Feb-99.pdf), accessed March 27, 2018.
* 195. EPA, “3-[N-Butyl-N-acetyl]-Aminopropionic Acid, Ethyl Ester (113509) Fact Sheet” (available at https://www3.epa.gov/pesticides/chem_search/reg_actions/registration/fs_PC-113509_01-Jan-00.pdf), accessed March 27, 2018.
* 196. FDA, “Food Additive Status List” (available at https://www.fda.gov/food/ingredientspackaginglabeling/foodadditivesingredients/ucm091048.htm), accessed March 27, 2018.
* 197. EPA, “Reregistration Eligibility Decision (R.E.D) Facts, Oil of Citronella,” February 1997 (available at https://www3.epa.gov/pesticides/chem_search/reg_actions/reregistration/fs_PC-021901_1-Feb-97.pdf), accessed March 27, 2018.
* 198. NPIC, “Oil of Citronella General Fact Sheet” (available at http://npic.orst.edu/factsheets/citronellagen.pdf), accessed March 27, 2018.
* 199. EPA, Office of Pesticide Programs, Label Review Manual, “Chapter 3: General Labeling Requirements,” revised March 2018 (available at https://www.epa.gov/sites/production/files/2017-10/documents/chap-03-dec-2014.pdf), accessed March 27, 2018.
* 200. CDC, Travelers' Health, Chapter 2: The Pretravel Consultation. J. Mutebi, W.A. Hawley, and W.G. Brogdon, “Protection against Mosquitoes, Ticks, and Other Arthropods” (available at https://wwwnc.cdc.gov/travel/yellowbook/2018/the-pre-travel-consultation/protection-against-mosquitoes-ticks-other-arthropods), accessed March 27, 2018.
201. American Academy of Pediatrics, “Tips for Using Repellents Safely” (available at www.healthychildren.org/English/safety-prevention/at-play/Pages/Insect-Repellents.aspx?rf), accessed March 27, 2018.
202. EPA, “DEET” (available at https://www.epa.gov/insect-repellents/deet#children), accessed November 28, 2018.
203. Kaldor, J., D. Shugg, B. Young, et al., “Non-Melanoma Skin Cancer: Ten Years of Cancer-Registry-Based Surveillance,” International Journal of Cancer, vol. 53, pp. 886-891, 1993.
204. Girschik, J., L. Fritschi, T. Threlfall, et al., “Deaths from Non-Melanoma Skin Cancer in Western Australia,” Cancer Causes and Control, vol. 19, pp. 879-885, 2008.
205. Ross, E.A., K.A. Savage, L.J. Utley, et al., “Insect Repellant Interactions: Sunscreens Enhance DEET (N,N-Diethyl-M-Toluamide) Absorption,” Drug Metabolism and Disposition, vol. 32, pp.783-785, 2004.
206. Gu, X., T. Wang, D.M. Collins, et al., “In Vitro Evaluation of Concurrent Use of Commercially Available Insect Repellent and Sunscreen Preparations,” British Journal of Dermatology, vol. 152(6), pp. 1263-1267, 2005.
207. Wang, T. and X. Gu, “In Vitro Percutaneous Permeation of the Repellent DEET and the Sunscreen Oxybenzone Across Human Skin,” Journal of Pharmacy and Pharmaceutical Sciences, vol. 10(1), pp. 17-25, 2007.
208. Kasichayanula, S., J.D. House, T. Wang, et al., “Simultaneous Analysis of Insect Repellent DEET, Sunscreen Oxybenzone and Five Relevant Metabolites by Reversed-Phase HPLC with UV Detection: Application to an In Vivo Study in a Piglet Model,” Journal of Chromatography B. Analytical Technologies in the Biomedical and Life Sciences, vol. 882(1-2), pp. 271-277, 2005.
209. Kasichayanula S., J.D. House, T. Wang, et al., “Percutaneous Characterization of the Insect Repellent DEET and the Sunscreen Oxybenzone from Topical Skin Application,” Toxicology and Applied Pharmacology, vol. 223(2), pp. 187-194, 2007.
210. Yinn, L.M., J.N. Tian, and C.C. Hung, “Assessment of Dermal Absorption of DEET-Containing Insect Repellent and Oxybenzone-Containing Sunscreen Using Human Urinary Metabolites,” Environmental Science and Pollution Research International, vol. 22, pp. 2062-2070, 2015.
211. Montemarano, A., R. Gupta, J. Burge, et al., “Insect Repellents and the Efficacy of Sunscreens,” The Lancet, vol. 349(9066), pp. 1670-1671, 1997.
Start List of SubjectsList of Subjects
21 CFR Part 201
- Drugs
- Incorporation by reference
- Labeling
- Reporting and recordkeeping requirements
21 CFR Part 310
- Administrative practice and procedure
- Drugs
- Labeling
- Medical devices
- Reporting and recordkeeping requirements
21 CFR Part 347
- Labeling
- Over-the-counter drugs
21 CFR Part 352
- Labeling
- Over-the-counter drugs
Therefore, under the Federal Food, Drug, and Cosmetic Act and under authority delegated to the Commissioner of Food and Drugs, we propose that 21 CFR parts 201, 310, 347, and 352 be amended as follows:
Start PartPART 201—LABELING
End Part Start Amendment Part1. The authority citation for part 201 continues to read as follows:
End Amendment Part Start Amendment Part2. Revise § 201.327 to read as follows:
End Amendment PartStart PartOver-the-counter sunscreen drug products; required labeling based on effectiveness testing.The following provisions apply to an over-the-counter (OTC) sunscreen drug product that is intended for application to the skin of humans for purposes of absorbing, reflecting, or scattering radiation in the ultraviolet (UV) range at wavelengths from 290 to 400 nanometers (nm), and that contains one or more of the following as an active ingredient: Avobenzone, cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, oxybenzone, padimate O, sulisobenzone, titanium dioxide, or zinc oxide, alone or in combination. The provisions do not apply to OTC sunscreen drug products marketed under approved new drug applications or abbreviated new drug applications. The failure of a product covered by this section to comply with any provision of this section, including the labeling of such a product with any effectiveness claim based on testing that fails to comply with any provision of this section, renders that product misbranded under section 502 of the Federal Food, Drug, and Cosmetic Act.
(a) General obligations of responsible persons. As used in this section, a “responsible person” is the manufacturer, packer, or distributor whose name appears on the labeling of a product covered by this section. A responsible person must assure that final formulation testing conducted on its product(s) pursuant to paragraphs (i) and (j) of this section complies with all applicable provisions of this section.
(1) Transfer of obligations. (i) A responsible person may transfer responsibility for any or all of its obligations set forth in this section to another entity (e.g., a contract research organization and/or testing laboratory), except as set forth in paragraph (l) (recordkeeping) of this section. Any such transfer must be described in writing. If not all obligations are transferred, the writing is required to describe each of the obligations being assumed by the transferee. If all obligations are transferred, a general statement that all obligations have been transferred is acceptable. Any obligation not covered by the written description will be deemed not to have been transferred. A written record of the transfer of obligations must be maintained by both parties to the transfer for the time period set forth in paragraph (l) of this section.
(ii) An entity that assumes any obligation(s) of a responsible person must comply with the provisions of this section applicable to the assumed obligation and will be subject to the same regulatory action as a responsible person for failure to comply with any obligation assumed under this section. Thus, all references to “responsible person” in this section apply to another entity (e.g., a contract research organization or testing laboratory) to the extent that it assumes one or more obligations of a responsible person.
(2) Personnel. A responsible person must select only investigators and other personnel qualified by appropriate training and/or experience to conduct final formulation testing pursuant to this section. Personnel engaged in testing under this section must have the education, training, and experience, or any combination thereof, to enable that person to adequately perform their assigned functions.
(b) Principal display panel. The following labeling must be prominently placed on the principal display panel:
(1) Statement of identity—(i) Placement. The principal display panel of an over-the-counter sunscreen drug product bears a statement of identity as one of its principal features. Except as set forth in paragraph (h) of this section, the statement of identity consists of the name of each sunscreen active ingredient in the product as identified in this section, listed in alphabetical order and followed by “Sunscreen” and “[Dosage form]” (e.g., “Lotion” “Spray”).
(ii) Prominence. The statement of identity must appear on the principal display panel in boldface type at least one-quarter as large as the size of the most prominent printed matter on the principal display panel, in lines generally parallel to the base on which the package rests as it is designed to be displayed and in direct conjunction with the most prominent display of the proprietary name or designation. The entire text of the statement of identity must appear in the same font style, size, and color with the same background color, and as continuous text with no intervening text or graphic, other than additional text provided in accordance with paragraph (h) of this section.
(2) Effectiveness claim. For purposes of this section, the term “determined SPF value” refers to the SPF value that equals the largest whole number less than SPF−(t*SE), determined for a sunscreen product in accordance with paragraph (i) of this section.
(i) SPF Broad Spectrum Statement. For a product that has been shown to pass the broad spectrum test in paragraph (j) of this section, the labeling states “Broad Spectrum SPF [insert the labeled SPF value associated with the range into which the determined SPF value falls, as set forth in the following table.]”
Table 1 to Paragraph (b)(2)(i)—SPF Labeling Ranges
Range of determined SPF values Associated labeled SPF value 60-80 60+. 50-59 50. 40-49 40. 30-39 30. 25-29 25. 20-24 20. 15-19 15. 2-14 Determined SPF Value. (ii) SPF Statement. For a product that has not been shown to pass the broad spectrum test in paragraph (j) of this section, the labeling states “SPF [insert labeled SPF value associated with the range into which the determined SPF value falls, as set forth in the table in paragraph (b)(2)(i) of this section]”.
(iii) For a product with a determined SPF value of at least 2 but less than 15. The SPF statement is immediately followed by an asterisk (“*”), and the associated statement “*See Skin Cancer/Skin Aging Alert” appears in the bottom 30 percent of the principal display panel.
(iv) Prominence of required statements. The SPF Broad Spectrum statement, SPF statement, and “*See Skin Cancer/Skin Aging Alert” statement, as applicable, must appear in boldface type at least one-quarter as large as the most prominent printed matter on the principal display panel and in lines generally parallel to the base on which the package rests as it is designed to be displayed. The entire text of the Broad Spectrum SPF or SPF statement, as applicable, must appear in the same font style, size, and color with the same background color and must appear as continuous text with no intervening text or graphic. The entire text of the “See Skin Cancer/Skin Aging Alert” statement, as applicable, must appear in the same font style, size, and color with the same background color and must appear as continuous text with no intervening text or graphic.
(3) Water resistance statements—(i) For products that provide 40 minutes of water resistance according to the test in paragraph (i)(8)(i) of this section. The Start Printed Page 6265labeling states “Water Resistant (40 minutes).”
(ii) For products that provide 80 minutes of water resistance according to the test in paragraph (i)(8)(ii) of this section. The labeling states “Water Resistant (80 minutes).”
(iii) Prominence of water resistance statement. For all products bearing a water resistance statement, the statement must appear in boldface type at least one-quarter as large as the most prominent printed matter on the principal display panel and in lines generally parallel to the base on which the package rests as it is designed to be displayed. The entire text of the water resistance statement must appear in the same font style, size, and color with the same background color, and as continuous text with no intervening text or graphic.
(c) Indications. The labeling of the product states, under the heading “Uses,” the phrases listed in this paragraph, as appropriate. Other truthful and nonmisleading statements, describing only the uses that have been established and listed in this paragraph, may also be used, as provided in § 330.1(c)(2) of this chapter, subject to the provisions of section 502 of the Federal Food, Drug, and Cosmetic Act relating to misbranding and the prohibition in section 301(d) of the Federal Food, Drug, and Cosmetic Act against the introduction or delivery for introduction into interstate commerce of unapproved new drugs in violation of section 505(a) of the Federal Food, Drug, and Cosmetic Act.
(1) For all sunscreen products, the following indication statement must be included under the heading “Uses”: “[bullet] helps prevent sunburn”. See § 201.66(b)(4) for definition of bullet.
(2) For sunscreen products that have been shown to pass the broad spectrum test in paragraph (j) of this section and have a determined SPF value of 15 or higher, the labeling may include the following statement in addition to the indication in paragraph (c)(1) of this section: “[bullet] if used as directed with other sun protection measures (see Directions [in bold italic font]), decreases the risk of skin cancer and early skin aging caused by the sun”.
(3) Any labeling or promotional materials that suggest or imply that the use, alone, of any sunscreen reduces the risk of or prevents skin cancer or early skin aging will cause the product to be misbranded under section 502 of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 352).
(d) Warnings. The labeling of the product contains the following warnings under the heading “Warnings”.
(1) For all sunscreen products. (i) The labeling states “Do not use [bullet] on damaged or broken skin.”
(ii) The labeling states “When using this product [bullet] keep out of eyes. Rinse with water to remove.”
(iii) The labeling states “Stop use and ask a doctor if [bullet] rash occurs.”
(2) For sunscreen products that are broad spectrum with determined SPF values of at least 2 but less than 15 according to the SPF test in paragraph (i) of this section or that have not been shown to pass the broad spectrum test in paragraph (j) of this section. The first statement under the heading “Warnings” states “Skin Cancer/Skin Aging Alert [in bold font]: Spending time in the sun increases your risk of skin cancer and early skin aging. This product has been shown only to help prevent sunburn, not [in bold font] skin cancer or early skin aging.”
(3) For products in a spray dosage form that meet the definition of either the term “flammable” or the term “combustible” as defined in § 352.3(g) or (h) of this chapter, as applicable, when tested in accordance with 16 CFR 1500.43a—(i) Labeling statement. The labeling states [bullet] “Flammable” or “Combustible” (as applicable) followed by a colon and the statement “Keep away from fire or flame.”
(ii) For products that have a drying time of less than 5 minutes. The labeling states [bullet] “Wait 5 minutes after application before approaching a source of heat or flame, or before smoking.”
(iii) For products that have a drying time of at least 5 minutes but less than 10 minutes. The labeling states [bullet] “Wait 10 minutes after application before approaching a source of heat or flame, or before smoking.”
(e) Directions. The labeling of the product contains the following statements, as appropriate, under the heading “Directions.” More detailed directions applicable to a particular product formulation may also be included.
(1) For all sunscreen products. (i) As an option, the labeling may state “For sunscreen use:”.
(ii) The labeling states “[bullet] apply [select one of the following: `liberally' or `generously'] [and, as an option: `and evenly'] 15 minutes before sun exposure”.
(iii) As an option, the labeling may state “[bullet] apply to all skin exposed to the sun”.
(iv) The labeling states “[bullet] children under 6 months of age: Ask a doctor”.
(2) For sunscreen products that have been shown to pass the broad spectrum test in paragraph (j) of this section and have a determined SPF value of 15 or higher. The labeling states “[bullet] Sun Protection Measures. [in bold font] Spending time in the sun increases your risk of skin cancer and early skin aging. To decrease this risk, regularly use a sunscreen with a Broad Spectrum SPF value of 15 or higher and other sun protection measures including: [bullet] limit time in the sun, especially from 10 a.m.-2 p.m. [bullet] wear long-sleeved shirts, pants, hats, and sunglasses”.
(3) For products that satisfy the water resistance test in paragraph (i)(8) of this section. The labeling states “[bullet] reapply: [bullet] after [select one of the following determined by water resistance test: ‘40 minutes of’ or ‘80 minutes of’] swimming or sweating [bullet] immediately after towel drying [bullet] at least every 2 hours”.
(4) For products that do not satisfy the water resistance test in paragraph (i)(8) of this section. The labeling states “[bullet] reapply at least every 2 hours [bullet] use a water resistant sunscreen if swimming or sweating”.
(5) For sunscreen products in a spray dosage form. The labeling states “[bullet] Hold container 4 to 6 inches from the skin to apply. [bullet] Do not spray directly into face. Spray on hands then apply to face. [bullet] Do not apply in windy conditions. [bullet] Use in a well-ventilated area and avoid inhalation”.
(f) Other information. The labeling of the product contains the following statement under the heading “Other information:” “[bullet] protect the product in this container from excessive heat and direct sun”.
(g) False or misleading claims. There are claims that would be false and/or misleading on sunscreen products. These claims include but are not limited to the following: “Sunblock,” “sweatproof,” and “waterproof.” These or similar claims will cause the product to be misbranded under section 502 of the Federal Food, Drug, and Cosmetic Act.
(h) Labeling of products containing a combination of sunscreen and skin protectant active ingredients. Statements of identity, indications, warnings, and directions for use, respectively, applicable to each ingredient in the product may be combined to eliminate duplicative words or phrases so that the resulting information is clear and understandable. Labeling provisions in § 347.50(e) of this chapter do not apply to these products.
(1) Statement of identity. The statement of identity of a sunscreen product that also contains one or more skin protectant active ingredients, identified in §§ 347.10(a), (d), (e), (g), h), Start Printed Page 6266(i), (k), (l), (m), and (r) of this chapter, consists of the names of all sunscreen and skin protectant active ingredients in alphabetical order followed by “Sunscreen/Skin Protectant” and “[Dosage form].” The statement of identity must be prominently placed on the principal display panel and presented in accordance with paragraph (b)(1)(ii) of this section.
(2) Indications. The labeling of the product states, under the heading “Uses,” any or all of the applicable indication(s) included in § 347.50(b) of this chapter or in paragraph (c) of this section. Other truthful and nonmisleading statements, describing only the indications for use that have been established in § 347.50(b) of this chapter or listed in paragraph (c) of this section, may also be used, as provided by § 330.1(c)(2) of this chapter, subject to the provisions of section 502 of the Federal Food, Drug, and Cosmetic Act relating to misbranding and the prohibition in section 301(d) of the Federal Food, Drug, and Cosmetic Act against the introduction or delivery for introduction into interstate commerce of unapproved new drugs in violation of section 505(a) of the Federal Food, Drug, and Cosmetic Act.
(3) Warnings. The labeling of the product states, under the heading “Warnings,” the applicable warnings for sunscreens in paragraph (d) of this section and for skin protectants in § 347.50(c) of this chapter.
(4) Directions. The labeling of the product states, under the heading “Directions,” any or all of the applicable directions for sunscreens, as set forth in paragraph (e) of this section, and for skin protectants, as set forth in §§ 347.50(d) and 347.60(d) of this chapter, unless otherwise stated in this paragraph. When the time intervals or age limitations for administration of the individual ingredients differ, the directions for the product may not contain any dosage that exceeds those established for any individual ingredient in the applicable OTC drug monograph(s), and may not provide for use by any age group lower than the highest minimum age limit established for any individual ingredient. When the directions for administration of the sunscreen and skin protectant differ in any other way, the directions for sunscreens in paragraph (e) of this section should be used.
(i) Sun Protection Factor (SPF) testing—(1) Adequate clinical testing procedures and conditions— (i) General obligations of responsible persons for testing under this paragraph. Responsible persons must provide investigators and other personnel engaged in SPF testing with the information they need to conduct an investigation properly; must obtain a signed investigator statement from each investigator; must ensure proper monitoring of the investigation(s); must ensure that the investigation(s) is conducted in accordance with written general investigational plan(s) and protocol(s); must ensure compliance with paragraphs (i)(1)(ii) and (iii) of this section; and must ensure that FDA and all participating investigators are promptly informed of significant new adverse effects or risks with respect to the drug.
(ii) Informed consent. Effective informed consent, as defined in part 50 of this chapter, must be obtained from all human subjects before initiating clinical final formulation testing under this paragraph (i).
(iii) Institutional review board (IRB) approval. Clinical testing under this paragraph (i), must be reviewed and approved by an IRB meeting the requirements of FDA's regulations in part 56 of this chapter.
(iv) Control of personnel—(A) General obligations. A responsible person is responsible for ensuring that investigators and other personnel conducting any testing under this paragraph (i), conduct all investigations in accordance with the signed investigator statement, the investigational plan, and applicable regulations. Responsible persons must ensure the implementation of adequate safeguards to protect the rights, safety, and welfare of subjects under he investigator's care. The responsible person must also ensure that investigators or other study personnel will promptly report to the IRB all changes in the clinical final formulation testing and all unanticipated problems involving risk to human subjects or others, and that investigators or other personnel will not make any changes in the clinical final formulation testing without IRB approval, except where necessary to eliminate apparent immediate hazards to human subjects.
(B) Obtaining information from the investigator. Before permitting an investigator to begin participating in clinical final formulation testing under this paragraph (i), the responsible person must obtain the following:
(1) Investigator statement. A signed investigator statement containing the name and address of the investigator and a commitment by the investigator that he or she—
(i) Will conduct the testing in accordance with the relevant, current protocol(s) and will only make changes in a protocol after notifying the responsible person and the IRB, except when necessary to protect the safety, the rights, or welfare of subjects;
(ii) Will comply with all requirements regarding the obligations of clinical investigators and all other pertinent requirements in this subpart;
(iii) Will personally conduct or supervise the described investigation(s);
(iv) Will inform any potential subjects that the drugs are being used for investigational purposes and will comply with the requirements relating to obtaining informed consent (part 50 of this chapter) and institutional review board review and approval (part 56 of this chapter);
(v) Will report to the responsible person adverse experiences that occur during the investigation(s);
(vi) Will ensure that all personnel assisting in the conduct of the testing are informed about their obligations in meeting the above commitments.
(2) Curriculum vitae. A curriculum vitae or other statement of qualifications of the investigator showing the education, training, and experience that qualifies the investigator to conduct the final formulation testing pursuant to this paragraph (i).
(v) Informing investigators. The responsible person must, as the overall investigation proceeds, keep each participating investigator informed of new observations discovered by or reported to the responsible person on the drug, particularly with respect to adverse effects and safe use.
(vi) Review of ongoing investigations. (A) The responsible person must monitor the progress of all clinical testing being conducted on its final formulation pursuant to this paragraph (i).
(B) A responsible person who discovers noncompliance by an investigator or other personnel with the signed agreement, the general investigational plan, or the requirements of this paragraph (i) or other applicable regulations (e.g., parts 50 and 56 of this chapter) must promptly either secure compliance or end the investigator's or other personnel's participation in testing conducted under this paragraph (i).
(C) The responsible person must review and evaluate the evidence relating to the safety and effectiveness of the final formulation as it is obtained from the investigator.
(vii) Investigator reports—(A) Safety reports. An investigator must immediately report to the responsible person any serious adverse event, whether or not considered related to the final formulation, including those listed in the protocol, and must include an Start Printed Page 6267assessment of whether there is a reasonable possibility that the final formulation being tested caused the adverse event. The investigator must record nonserious adverse events and report them to the responsible person according to the timetable specified in the protocol.
(B) Final report. An investigator must provide the responsible person with an adequate report shortly after completion of each investigation conducted by that investigator for the responsible person under this paragraph (i).
(2) UV source (solar simulator)—(i) Emission spectrum. Filter a single port or multiport solar simulator so that it provides a continuous emission spectrum from 290 to 400 nanometers (nm) with a limit of 1,500 watts per square meter (W/m2) on total irradiance for all wavelengths between 250 and 1,400 nm.
(A) The solar simulator must have the following percentage of erythema-effective radiation in each specified range of wavelengths:
Table 2 to Paragraph (i)(2)(i)(A)—Solar Simulator Emission Spectrum
Wavelength range (nm) Percent erythemal contribution 1 <290 <0.1 290-300 1.0-8.0 290-310 49.0-65.0 290-320 85.0-90.0 290-330 91.5-95.5 290-340 94.0-97.0 290-400 99.9-100.0 1 Calculation of erythema action spectrum described in paragraph (i)(2)(ii) of this section. (B) In addition, UVA II (320-340 nm) irradiance must equal or exceed 20 percent of the total UV (290-400 nm) irradiance. UVA I (340-400 nm) irradiance must equal or exceed 60 percent of the total UV irradiance.
(ii) Erythema action spectrum. (A) Calculate the erythema action spectrum weighting factor (Vi) at each wavelength λ:
(1) Vi (λ) = 1.0 (250 <λ ≤298 nm)
(2) Vi (λ) = 100.094 * (298 − λ) (298 <λ ≤328 nm)
(3) Vi (λ) = 100.015 * (140 − λ) (328 <λ ≤400 nm)
(B) Calculate the erythema-effective UV dose (E) delivered by a solar simulator as follows:

Where
Vi (λ) = erythema action spectrum weighting factor at each wavelength λ
I(λ) = irradiance (Watts per square meter) at each wavelength λ
t = exposure time (seconds)
Erythema-effective dose (E) is expressed as effective Joules per square meter (J/m2-eff).
(C) The solar simulator radiation intensity must be determined using a handheld radiometer with a response weighted to match the spectrum in ISO 17166 CIE S 007/E entitled “Erythemal reference action spectrum and standard erythema dose,” dated 1999 (First edition, 1999-12-15; corrected and reprinted 2000-11-15), which is incorporated by reference in accordance with 5 U.S.C. 552(a) and 1 CFR part 51. You may obtain a copy from the ISO Copyright Office, Case Postale 56, CH-1211, Geneva 20, Switzerland, telephone +41-22-749-01-11 or fax +41-22-74-09-47. https://www.iso.org. You may inspect a copy at the Center for Drug Evaluation and Research, 10903 New Hampshire Ave., Bldg. 22, Silver Spring, MD 20993, call 301-796-2090, or at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: https://www.archives.gov/federal-register/cfr/ibr-locations.html. The solar simulator output should be measured before and after each phototest or, at a minimum, at the beginning and end of each test day. This radiometer should be calibrated using side-by-side comparison with the spectroradiometer (using the weighting factors determined according to paragraph (i)(2)(ii)(A) of this section) at the time of the annual spectroradiometric measurement of the solar simulator as described in paragraph (i)(2)(iv) of this section.
(iii) Operation. A solar simulator must have no significant time-related fluctuations (within 20 percent) in radiation emissions after an appropriate warm-up time and demonstrate good beam uniformity (within 20 percent) in the exposure plane. The delivered dose to the UV exposure site must be within 10 percent of the expected dose.
(iv) Periodic measurement. To ensure that the solar simulator delivers the appropriate spectrum of UV radiation, the emission spectrum of the solar simulator must be measured at least annually with an appropriate and accurately calibrated spectroradiometer system (results should be traceable to the National Institute for Standards and Technology). In addition, the solar simulator must be recalibrated if there is any change in the lamp bulb or the optical filtering components (i.e., filters, mirrors, lenses, collimating devices, or focusing devices). Daily solar simulator radiation intensity should be monitored with a broadband radiometer with a response weighted to match the erythema action spectrum in ISO 17166 CIE S 007/E entitled “Erythemal reference action spectrum and standard erythema dose,” which is incorporated by reference in paragraph (i)(2)(ii)(C) of this section. If a lamp must be replaced due to failure or aging during a phototest, broadband device readings consistent with those obtained for the original calibrated lamp will suffice until measurements can be performed with the spectroradiometer at the earliest possible opportunity.
(3) SPF standard—(i) Preparation. The SPF standard must be a formulation containing 7-percent padimate O and 3-percent oxybenzone.
Table 3 to Paragraph (i)(3)(i)—Composition of the Padimate O/Oxybenzone SPF Standard
Ingredients Percent by weight Part A: Lanolin 4.50 Cocoa butter 2.00 Glyceryl monostearate 3.00 Stearic acid 2.00 Padimate O 7.00 Oxybenzone 3.00 Part B: Purified water USP 71.60 Sorbitol solution 5.00 Triethanolamine, 99 percent 1.00 Methylparaben 0.30 Propylparaben 0.10 Part C: Benzyl alcohol 0.50 Part D: Purified water USP QS 1 1 Quantity sufficient to make 100 grams. (A) Step 1. Add the ingredients of Part A into a suitable stainless steel kettle equipped with a propeller agitator. Mix at 77 to 82 °C until uniform.
(B) Step 2. Add the water of Part B into a suitable stainless steel kettle equipped with a propeller agitator and begin mixing at 77 to 82 °C. Add the remaining ingredients of Part B and mix until uniform.
(C) Step 3. Add the batch of Step 1 to the batch of Step 2 and mix at 77 to 82 °C until smooth and uniform. Slowly cool the batch to 49 to 54 °C.
(D) Step 4. Add the benzyl alcohol of Part C to the batch of Step 3 at 49 to 54 °C. Mix until uniform. Continue to cool batch to 35 to 41 °C.
(E) Step 5. Add sufficient water of Part D to the batch of Step 4 at 35 to 41 °C to obtain 100 grams of SPF standard. Mix until uniform. Cool batch to 27 to 32 °C.Start Printed Page 6268
(ii) HPLC assay. Use the following high performance liquid chromatography (HPLC) procedure to verify the concentrations of padimate O and oxybenzone in the SPF standard:
(A) Instrumentation—(1) Equilibrate a suitable liquid chromatograph to the following or equivalent conditions:
(i) Column C-18, 250 millimeters (mm) length, 4.6 mm inner diameter (5 microns). (ii) Mobile Phase 85:15:0.5 methanol: water: acetic acid. (iii) Flow Rate 1.5 milliliters (mL) per minute. (iv) Temperature Ambient. (v) Detector UV spectrophotometer at 308 nanometers. (vi) Attenuation As needed. (2) Use HPLC grade reagents for mobile phase.
(B) Preparation of the HPLC reference standard. (1) Weigh 0.5 gram (g) of oxybenzone USP reference standard into a 250-mL volumetric flask. Dissolve and dilute to volume with isopropanol. Mix well.
(2) Weigh 0.5 g of padimate O USP reference standard into a 250-mL volumetric flask. Dissolve and dilute to volume with isopropanol. Mix well.
(3) Pipet 3 mL of the oxybenzone solution and 7 mL of the padimate O solution into a 100-mL volumetric flask. Dilute to volume with isopropanol and mix well.
(C) HPLC system suitability. (1) Make three replicate 10-microliter injections of the HPLC reference standard (described in paragraph (i)(3)(ii)(B) of this section). The relative standard deviation in peak areas should not be more than 2 percent for either oxybenzone or padimate O.
(2) Calculate the resolution (R) between the oxybenzone and padimate O peaks from one chromatogram as follows:

Where
to = retention time for oxybenzone
tp = retention time for padimate O
Wo = oxybenzone peak width at baseline
Wp = padimate O peak width at baseline
If the resolution (R) is less than 3, adjust the mobile phase or replace the column.
(D) SPF standard assay. (1) The SPF standard is diluted to the same concentration as the HPLC reference standard according to the following steps:
(i) Step 1. Weigh 1 g of the SPF standard (described in paragraph (i)(3)(i) of this section) into a 50-mL volumetric flask.
(ii) Step 2. Add approximately 30 mL of isopropanol and heat with swirling until contents are evenly dispersed.
(iii) Step 3. Cool to room temperature (15 to 30 °C) and dilute to volume with isopropanol. Mix well.
(iv) Step 4. Pipet 5.0 mL of the preparation into a 50-mL volumetric flask and dilute to volume with isopropanol. Mix well.
(2)(i) Inject 10-microliter of diluted SPF standard from paragraph (i)(3)(ii)(D)(1) of this section and calculate the amount of oxybenzone and padimate O as follows:

(ii) The percent of oxybenzone and padimate O in the SPF standard must be between 95 and 105.
(4) Test subjects—(i) Number of subjects. A test panel should include enough subjects to produce a minimum of 10 valid test results. A maximum of three subjects may be rejected from this panel based on paragraph (i)(6)(v) of this section.
(ii) Medical history. (A) Obtain a medical history from each subject with emphasis on the effects of sunlight on the subject's skin. Determine that each subject is in good general health with skin type I, II, or III as follows:
(1) Always burns easily; never tans (sensitive).
(2) Always burns easily; tans minimally (sensitive).
(3) Burns moderately; tans gradually (light brown) (normal).
(4) Burns minimally; always tans well (moderate brown) (normal).
(5) Rarely burns; tans profusely (dark brown) (insensitive).
(6) Never burns; deeply pigmented (insensitive).
(B) Skin type is based on first 30 to 45 minutes of sun exposure after a winter season of no sun exposure. Determine that each subject is not taking topical or systemic medication that is known to alter responses to UV radiation. Determine that each subject has no history of sensitivities to topical products and/or abnormal responses to sunlight, such as a phototoxic or photoallergic response.
(iii) Physical examination. Conduct a physical examination to determine the presence of sunburn, suntan, scars, active dermal lesions, and uneven skin tones on the areas of the back to be tested. Adequate time must have passed following any previous UV exposure (e.g., participation in a prior SPF clinical study, tanning, etc.) so that the test subject has no preexisting skin pigmentation at the time of enrollment. A suitable source of low power UVA, such as a Woods lamp, is helpful in this process. If any of these conditions are present, the subject is not qualified to participate in the study. The presence of nevi, blemishes, or moles will be acceptable if, in the physician's judgment, they will neither compromise the study nor jeopardize a subject's safety. Subjects with dysplastic nevi should not be enrolled. Excess hair on the back is acceptable if the hair is clipped. Shaving is unacceptable because it may remove a significant portion of the stratum corneum and temporarily alter the skin's response to UV radiation.
(iv) Informed consent. Obtain legally effective written informed consent from all test subjects as required by paragraph (i)(1)(ii) of this section.Start Printed Page 6269
(5) Sunscreen application—(i) Test site. Test sites are locations on each subject's back, between the beltline and the shoulder blades (scapulae) and lateral to the midline, where skin responses to UV radiation are determined. Responses on unprotected skin (no test material applied) and protected skin (sunscreen test product(s) or SPF standard applied) are determined at separate unprotected and protected test sites, respectively. Test sites should be randomly located in a blinded manner. Each test site should be a minimum of 30 square centimeters and outlined with indelible ink.
(ii) Test subsite. Test subsites are the locations to which UV radiation is administered within a test site. Administer UV doses to at least five test subsites within each test site. Test subsites must be at least 0.5 square centimeters (cm2) in area and must be separated from each other by at least 0.8 cm. Each test subsite must be outlined with indelible ink.
(iii) Applying test materials. Apply the sunscreen test product and the SPF standard at 2 milligrams per square centimeter (mg/cm2) to their respective test sites. Use a finger cot compatible with the sunscreen to spread the product as evenly as possible.
(iv) Waiting period. Wait at least 15 minutes after applying a sunscreen product before exposing the test sites to UV radiation as described in paragraph (i)(6) of this section. For water resistant sunscreen products, proceed with the water resistance testing procedure described in paragraph (i)(8) of this section after waiting at least 15 minutes.
(6) UV exposure and erythema reading—(i) Definition of minimal erythema dose (MED). The minimal erythema dose (MED) is the smallest UV dose (quantity of erythema-effective energy expressed as Joules per square meter) that produces perceptible redness of the skin (erythema) with clearly defined borders at 16 to 24 hours after UV exposure. The MED for unprotected skin (MEDu) is determined on a test site that does not have sunscreen applied. The MED for protected skin (MEDp) is determined on a test site that has sunscreen applied. An MEDp is determined for the SPF standard (ssMEDp). An MEDp is determined for the sunscreen test product (tpMEDp).
(ii) UV exposure for initial MEDu. For each test subject, no more than 1 day before testing a product, determine the initial MEDu by administering a series of UV radiation doses expressed as J/m2-eff (as determined according to paragraph (i)(2)(ii)(B) of this section) to the test subsites within an unprotected test site using an accurately calibrated solar simulator. Select doses that are a geometric series represented by 1.25n (i.e., each dose is 25 percent greater than the previous dose).
(iii) UV exposure for final MEDu,ss MEDp, andtp MEDp. For each subject, determine the final MEDu,ss MEDp, andtp MEDp by administering a series of five UV doses to the appropriate test sites. The middle dose (X) in each of these dose series (i.e., the third dose) should equal the initial MEDu times the expected SPF. Note that the expected SPF equals 1 and 16.3 for the final MEDu and ssMEDp, respectively. The remaining UV doses in the series depend upon the expected SPF value of the sunscreen test product(s). For products with an expected SPF less than 8, administer UV doses that increase by 25 percent with each successive dose (i.e., 0.64X, 0.80X, 1.00X, 1.25X, and 1.56X). For products with an expected SPF from 8 to 15, administer UV doses that increase by 20 percent with each successive dose (i.e., 0.69X, 0.83X, 1.00X, 1.20X, and 1.44X). For products with an expected SPF higher than 15, administer UV doses that increase by 15 percent with each successive dose (i.e., 0.76X, 0.87X, 1.00X, 1.15X, and 1.32X).
(iv) Evaluation of test subsites. In order that the study personnel who evaluates the test subsites is not biased, he/she should not be the same study personnel who applied the sunscreen product to the test site or administered the UV doses. After UV doses are administered, record all immediate responses. These may include an immediate darkening or tanning, typically grayish or purplish in color, which fades in 30 to 60 minutes; an immediate reddening at the subsite, due to heating of the skin, which fades rapidly; and an immediate generalized heat response, spreading beyond the subsite, which fades in 30 to 60 minutes. After the immediate responses are noted, each subject should shield the exposed area from further UV radiation until the MED is determined. Determine the final MEDu,ss MEDp, andtp MEDp 16 to 24 hours after UV exposure. Evaluate the erythema responses of each test subsite using either tungsten or warm white fluorescent lighting that provides at least 450 lux of illumination at the test site. For the evaluation, the test subject should be in the same position as when the test site was irradiated.
(v) Invalid test data. Reject test data for a test subject if erythema is not present on either the unprotected or protected test sites; or erythema is present at all subsites; or the responses are inconsistent with the series of UV doses administered; or the subject was noncompliant (e.g., the subject withdraws from the test due to illness or work conflicts or does not shield the exposed testing sites from further UV radiation until the MED is determined).
(7) Determination of SPF. (i) Calculate an SPF value for each test subject (SPFi) as follows:

(ii) Calculate the mean

and the standard deviation(s) from the SPFi values. Calculate the standard error (SE), which equals s/√n (where n equals the number of subjects who provided valid test results). Obtain the t value from Student's t distribution table corresponding to the upper 5-percent point with n − 1 degrees of freedom. Determine the SPF value that is equal to the largest whole number less than

In order for the SPF determination of a test product to be considered valid, the SPF value of the SPF standard must fall within the standard deviation range of the expected SPF (i.e., 16.3 ± 3.43).
(8) Determination of water resistance. To support labeling claims of water resistance in accordance with paragraph (b) of this section, the following procedure must be performed in an indoor fresh water pool, whirlpool, and/or hot tub maintained at 23 to 32 °C. Fresh water is clean drinking water that meets the standards in 40 CFR part 141. The pool and air temperature and the relative humidity must be recorded.
(i) Water resistance (40 minutes). Determine the SPF value after 40 minutes of water immersion using the following procedure:
(A) Step 1: Apply the sunscreen test product as described in paragraph (i)(5) of this section.
(B) Step 2: Perform moderate activity in water for 20 minutes.
(C) Step 3: Rest out of water for 15 minutes. Do not towel test site(s).
(D) Step 4: Perform moderate activity in water for 20 minutes.
(E) Step 5: Allow test sites to dry completely without toweling.
(F) Step 6: Apply the SPF standard as described in paragraph (i)(5) of this section.
(G) Step 7: Expose test sites to UV doses as described in paragraph (i)(6) of this section.
(ii) Water resistance (80 minutes). Determine the SPF value after 80 minutes of water immersion using the following procedure:Start Printed Page 6270
(A) Step 1: Apply the sunscreen test product as described in paragraph (i)(5) of this section.
(B) Step 2: Perform moderate activity in water for 20 minutes.
(C) Step 3: Rest out of water for 15 minutes. Do not towel test site(s).
(D) Step 4: Perform moderate activity in water for 20 minutes.
(E) Step 5: Rest out of water for 15 minutes. Do not towel test site(s).
(F) Step 6: Perform moderate activity in water for 20 minutes.
(G) Step 7: Rest out of water for 15 minutes. Do not towel test site(s).
(H) Step 8: Perform moderate activity in water for 20 minutes.
(I) Step 9: Allow test sites to dry completely without toweling.
(J) Step 10: Apply the SPF standard as described in paragraph (i)(5) of this section.
(K) Step 11: Expose test sites to UV doses as described in paragraph (i)(6) of this section.
(j) Broad spectrum testing— (1) UV Spectrometry— (i) Plate. Use optical-grade polymethylmethacrylate (PMMA) plates suitable for UV transmittance measurements. The plate should be roughened on one side to a three-dimensional surface topography measure (Sa) between 2 and 7 micrometers and must have a rectangular application area of at least 16 square centimeters (with no side shorter than 4 cm).
(ii) Sample holder. The sample holder should hold the PMMA plate in a horizontal position to avoid flowing of the sunscreen product from one edge of the PMMA plate to the other. Mount the PMMA plate as close as possible to the input optics of the spectrometer to maximize capture of forward scattered radiation. The sample holder should be a thin, flat plate with a suitable aperture through which UV radiation can pass. Place the PMMA plate on the upper surface of the sample holder with the roughened side facing up.
(iii) Light source. The light source must produce a continuous spectral distribution of UV radiation from 290 to 400 nanometers.
(iv) Input optics. Unless the spectrometer is equipped with an integrating sphere, an ultraviolet radiation diffuser should be placed between the sample and the input optics of the spectrometer. The diffuser will be constructed from any UV radiation transparent material (e.g., Teflon or quartz). The diffuser ensures that the radiation received by the spectrometer is not collimated. Set the spectrometer input slits to provide a bandwidth that is less than or equal to 5 nanometers.
(v) Dynamic range of the spectrometer. The dynamic range of the spectrometer should be sufficient to measure transmittance accurately through a highly absorbing sunscreen product at all terrestrial solar UV wavelengths (290 to 400 nm).
(2) Sunscreen product application to PMMA plate. The accuracy of the test depends upon the application of a precisely controlled amount of sunscreen product with a uniform distribution over the PMMA plate. The product is applied at 0.75 mg per square centimeter to the roughened side of the PMMA plate. The sunscreen product should be applied in a series of small amounts over the entire PMMA plate and then spread evenly using a gloved finger. Spreading should be done with a very light spreading action for approximately 30 seconds followed by spreading with greater pressure for approximately 30 seconds. The plate should then be allowed to equilibrate for 15 minutes in the dark before the pre-irradiation described in paragraph (j)(3) of this section.
(3) Sunscreen product pre-irradiation. To account for lack of photostability, irradiate the PMMA plate with a solar simulator described paragraph (i)(2) of this section. The irradiation dose must be 4 MEDs which is equivalent to an erythemal effective dose of 800 J/m2 (i.e., 800 J/m2-eff).
(4) Calculation of mean transmittance values. (i) After pre-irradiation, determine the mean transmittance values for each wavelength λ over the full UV spectrum (290 to 400 nanometers). Measure the transmittance values at 1 nanometer intervals. Measurements of spectral irradiance transmitted for each wavelength λ through control PMMA plates coated with 15 microliters of glycerin (no sunscreen product) must be obtained from at least five different locations on the PMMA plate [C1(λ), C2(λ), C3(λ), C4(λ), and C5(λ)]. In addition, a minimum of five measurements of spectral irradiance transmitted for each wavelength λ through the PMMA plate covered with the sunscreen product will be similarly obtained after pre-irradiation of the sunscreen product [P1(λ), P2(λ), P3(λ), P4(λ), and P5(λ)].
(ii) The mean transmittance for each wavelength is the ratio of the mean of the C(λ) values to the mean of the P(λ) values, as follows:

Where
n ≥5
(5) Calculation of mean absorbance values. (i) Mean transmittance values,

are converted into mean absorbance values,
at each wavelength by taking the negative logarithm of the mean transmittance value as follows:

(ii) The calculation yields 111 monochromatic absorbance values in 1 nanometer increments from 290 to 400 nanometers.
(6) Number of plates. For each sunscreen product, determine mean absorbance values from at least three individual PMMA plates. Because paragraph (j)(4) of this section requires at least 5 measurements per plate, there must be a total of at least 15 measurements.
(7) Calculation of the critical wavelength. The critical wavelength is identified as the wavelength at which the integral of the spectral absorbance curve reaches 90 percent of the integral over the UV spectrum from 290 to 400 nm. The following equation defines the critical wavelength:

Where
λc = critical wavelength
A(λ) = mean absorbance at each wavelength
dλ = wavelength interval between measurements
(8) Calculation of the UVA I/UV ratio. The ratio of UVA I/UV is calculated as the area (per unit wavelength) under the UVA I portions of a plot of wavelength versus A(λ), divided by the area (per unit wavelength) under the total curve, as follows:

Where
A(λ) = effective absorbance given as -log T(λ)mean absorbance at each wavelength,
d(λ) = wavelength interval between measurements
B(λ) = any biological action spectrum factor
Because no appropriate biological action spectrum for UVA radiation damage has been universally accepted, no action spectrum is specified. The value of B(λ) is, therefore, equal to 1.0 for all wavelengths.
(9) Determination of broad spectrum protection. A product that has both a mean critical wavelength of 370 nm or Start Printed Page 6271greater, calculated in accordance with paragraph (j)(7) of this section, and a mean UVA I/UV ratio of 0.70 or greater, calculated in accordance with paragraph (j)(8) of this section, is determined to pass the broad spectrum test.
(k) Regulatory status of final formulation testing and related requirements. Final formulation testing required under this section is considered a part of the manufacture of a sunscreen product. Therefore, final formulation testing required under this section must be performed in an establishment registered in accordance with part 207 of this chapter and section 510 of the Federal Food, Drug, and Cosmetic Act. Entities conducting final formulation testing required by this section must also comply with current good manufacturing practices (CGMPs) and associated recordkeeping requirements including those set forth in paragraph (l) of this section and in parts 210 and 211 of this chapter. Failure to comply with CGMPs or recordkeeping requirements will mean that any product labeled in reliance on that testing will be adulterated.
(l) Recordkeeping. Records required to be kept under this section must be maintained for at least 1 year after the expiration date of all products labeled in reliance on that testing or, in the case of certain OTC drug products lacking expiration dating because they meet the criteria for exemption under § 211.137 of this chapter, 3 years after distribution of the last lot of drug product bearing labeling that relies on the testing. Recordkeeping requirements under this section may not be transferred. Maintenance records required to be kept under (l)(1) must be kept by the testing entity. Records of final formulation testing as described in paragraphs (l)(2) and (3) of this section must be kept by the responsible person and any entity that is performing final formulation testing required by this section on behalf of a responsible person pursuant to a transfer of obligations.
(1) Maintenance records. Entities performing final formulation testing are expected to maintain equipment in accordance with paragraph (k) of this section and, as applicable, parts 210 and 211 of this chapter. Maintenance records must be kept for all equipment used for final formulation testing under this section and must include:
(i) Documentation that equipment has been maintained in accordance with established written specifications as required by paragraph (k) of this section and parts 210 and 211 of this chapter; and
(ii) Documentation of characterization of UV sources including:
(A) Record of emission spectrum, total irradiance, and percent of erythema-effective radiation contribution required by paragraph (i)(2)(i) of this section;
(B) Record of each periodic measurement required by paragraph (i)(2)(iv) of this section for each solar simulator;
(C) Record of each calibration, realignment, or change in components of each solar simulator, or any changes to the broadband radiometer (or UV meter/dose control system), required by paragraph (i)(2)(iv) of this section; and
(D) Record of each solar simulator output measurement required by paragraph (i)(2)(ii)(C) of this section.
(2) SPF testing records. In addition to any records required to be kept pursuant to parts 210 and 211 of this chapter, records of SPF testing performed pursuant to paragraph (i) of this section must include:
(i) Identification of the entity that conducted the final formulation testing, including the name and address of the establishment(s) at which testing was carried out;
(ii) The sunscreen test product identifier and characterization of the formulation being tested, including lot number, manufacture date, and expected SPF;
(iii) Characterization of the SPF standard sunscreen required by paragraph (i)(3) of this section, including:
(A) Lot number;
(B) Manufacturing date; and
(C) Results of HPLC SPF standard assay that verify compliance with the concentrations of padimate O and oxybenzone in the SPF standard.
(iv) Documentation linking any blinded samples with the product identifier.
(v) For each human subject, records of:
(A) The identification of the UV source used for testing on that subject, including make, model, and serial number;
(B) Initial and final individual MED for unprotected skin (MEDu), and the identity of the study personnel who determined that value;
(C) Final MED for sunscreen test product protected skin (tp MEDp), and the identity of the study personnel who determined that value;
(D) Final MED for SPF standard sunscreen protected skin (ss MEDp), and the identity of the study personnel who determined that value; and
(E) Individual SPFi values, including all valid test data and invalid test data for the test product and for the SPF standard sunscreen, and the identity of the study personnel who determined that value.
(vi) Records of the mean and standard deviation from SPFi values, standard error, and determined SPF value derived as set forth in paragraph (i)(7) of this section.
(vii) Records for water resistance testing of pool temperature, air temperature, and relative humidity as required by paragraph (i)(8) of this section.
(viii) Records demonstrating compliance with paragraph (i)(1) of this section governing the establishment of adequate clinical testing procedures and conditions, including, but not limited to:
(A) Case histories. Responsible persons are required to prepare and maintain adequate and accurate case histories on each individual participant enrolled in SPF testing performed under paragraph (i) of this section. Case histories must record all observations and other data pertinent to the investigation. Case histories include the case report forms and supporting data (for example, signed and dated consent forms, medical records including progress notes of the physician, the individual's hospital chart(s), and the nurses' notes (if applicable)). The case history for each individual participant must document that informed consent was obtained pursuant to part 50 before each individual's participation in the study. Case histories as required by this section must include:
(1) Protocol deviations or injuries, if any; and
(2) Identification, by subject, of the study personnel who: Examined the potential study site areas, who weighed and applied the sunscreen, and who provided the UV irradiation.
(B) IRB review. Documentation that clinical research conducted pursuant to paragraph (i) of this section was reviewed and approved by a registered IRB as required by paragraph (i)(1)(iii) of this section.
(3) Broad spectrum testing records. Records of broad spectrum testing conducted pursuant to paragraph (j) of this section must include:
(i) Identification of the entity that conducted the final formulation testing, including the name and address of the establishment(s) at which testing was carried out;
(ii) Records of sample information, including:
(A) A sunscreen test product identifier and expected SPF. If the samples used in testing under paragraph (j) of this section are blinded, then records must include a master key that enables samples to be re-identified. In all other cases, records must include a Start Printed Page 6272master key that links samples used to a sunscreen test product identifier.
(B) Sample number;
(C) Identifier code;
(D) Measurement of PMMA plate surface topography in micrometers; and
(E) Sample holder orientation (vertical or horizontal).
(iii) Identification of each UV source used for sunscreen product pre-irradiation, including make, model, and serial number.
(iv) Records of sunscreen product application, including:
(A) A record of all sample weights, including analytical balance; and
(B) For all equipment used; make, model, and serial number;
(v) For each sample, all measurements required by paragraphs (j)(4) to (6) of this section.
(vi) For each sample, records of critical wavelength and the UVA I/UV ratio values required by paragraphs (j)(7) and (8) of this section.
(vii) For each sample: The identity of the study personnel who weighed and applied the sunscreen to the PMMA plates; the identity of the study personnel who provided the pre-irradiation; and the identity of the study personnel, or, if calculated by software, what software, calculated the mean transmittance, mean absorbance values, critical wavelength, and UVA I/UV.
(viii) For each sample, the test dates for the broad spectrum test conducted pursuant to paragraph (j) of this section, and sample report forms and supporting data including, for example, spectral data, Excel files containing transmittance or absorbance values, or any notes from the lab investigator.
(4) Food and Drug Administration (FDA) inspection of records—(i) Testing entity. An entity performing final formulation testing under this section, including a responsible person or an entity that has been transferred any obligations of a responsible person under this section, must, upon request from any properly authorized officer or employee of FDA, at reasonable times, permit such officer or employee to have access to, and copy and verify any records or reports of testing pursuant to this section. The testing entity is not required to divulge subject names unless the records of particular individuals require a more detailed study of the cases, or unless there is reason to believe that the records do not represent actual case studies, or do not represent actual results obtained.
(ii) Responsible persons. A responsible person must upon request from any properly authorized officer or employee of FDA, at reasonable times, permit such officer or employee to have access to and copy and verify any records and reports relating to final formulation testing conducted under this section. Upon written request by FDA, the responsible person must submit the records or reports (or copies of them) to FDA. The responsible person must discontinue from further participation in final formulation testing required by this section any investigator who has failed to maintain or make available records or reports of the investigation as required by this paragraph (l).
PART 310—NEW DRUGS
End Part Start Amendment Part3. The authority citation for part 310 continues to read as follows:
End Amendment PartStart Amendment Part[Amended]4. Amend § 310.545 by removing and reserving paragraphs (a)(29) and (d)(31),and (40).
End Amendment Part Start Amendment Part5. Add § 310.549 to subpart E to read as follows:
End Amendment PartStart PartDrug products offered over-the-counter (OTC) for use as sunscreen.(a) Any drug product offered OTC for use as sunscreen and identified in any of paragraphs (b) through (i) of this section is not generally recognized as safe and effective and is regarded as a new drug within the meaning of section 201(p) of the Federal Food, Drug, and Cosmetic Act, for which an approved new drug application under section 505 of the Federal Food, Drug, and Cosmetic Act and part 314 of this chapter is required for marketing. In the absence of an approved new drug application, such product is also misbranded under section 502 of the Federal Food, Drug, and Cosmetic Act. Products offered OTC for use as sunscreen include those represented, labeled, or promoted as sunscreen, or for use to help prevent sunburn, skin cancer, and/or skin aging caused by the sun, or with similar claims or representations. Clinical investigations designed to obtain evidence that any sunscreen drug product covered by this section is safe and effective for the purpose intended must comply with the requirements and procedures governing the use of investigational new drugs set forth in part 312 of this chapter.
(b) A sunscreen drug product that contains any of the following ingredients:
(1) Diethanolamine methoxycinnamate
(2) Digalloyl trioleate
(3) Ethyl 4-[bis(hydroxypropyl)] aminobenzoate
(4) Glyceryl aminobenzoate
(5) Lawsone with dihydroxyacetone
(6) Red petrolatum
(7) Trolamine salicylate
(8) Aminobenzoic acid
(9) Avobenzone
(10) Cinoxate
(11) Dioxybenzone
(12) Ensulizole
(13) Homosalate
(14) Meradimate
(15) Octinoxate
(16) Octisalate
(17) Octocrylene
(18) Oxybenzone
(19) Padimate O
(20) Sulisobenzone
(c) A sunscreen drug product that has a determined sun protection factor (SPF) value, as defined in § 352.3(d) of this chapter, of at least 15 when tested in accordance with § 201.327(i) of this chapter, but that has not been shown to pass the broad spectrum test in § 201.327(j) of this chapter.
(d) A sunscreen drug product that has a determined sun protection factor (SPF) value, as defined in § 352.3(d) of this chapter, of less than 2 or greater than 80 when tested in accordance with § 201.327(i) of this chapter.
(e) A sunscreen drug product that has a determined sun protection factor (SPF) value, as defined in § 352.3(d) of this chapter, of less than 15 when tested in accordance with § 201.327(i) of this chapter and/or that does not pass the broad spectrum test in § 201.327(j) of this chapter, and labeled with any of the following or similar claims:
(1) Decreases the risk of skin cancer caused by the sun; or
(2) Decreases the risk of early skin aging caused by the sun.
(f) A sunscreen drug product labeled with any of the following or similar claims:
(1) Instant protection or protection immediately upon application; or
(2) Claims for “all-day” protection or extended wear claims citing a specific number of hours of protection that is inconsistent with the directions for application in § 201.327 of this chapter.
(g) A sunscreen drug product that is labeled, represented, or promoted for use as a combined sunscreen-insect repellant.
(h) A sunscreen drug product that is in any dosage form other than the following: Oil, lotion, cream, gel, butter, paste, ointment, stick, or spray.
(i) A sunscreen drug product in a spray dosage form that has any of the following properties:
(1) The product meets the definition of the term “extremely flammable” as Start Printed Page 6273defined at § 352.3(f) of this chapter when tested in accordance with 16 CFR 1500.43a;
(2) More than 10 percent of the particles dispensed from the consumer container are smaller than 10 micrometers;
(3) Any of the particles dispensed from the consumer container are smaller than 5 micrometers; or
(4) The product meets the definition of either the term “flammable” or the term “combustible” as defined at §§ 352.3(g) or (h) of this chapter, as applicable, when tested in accordance with 16 CFR 1500.43a and has a measured drying time of 10 minutes or more.
PART 347—SKIN PROTECTANT DRUG PRODUCTS FOR OVER-THE-COUNTER HUMAN USE
End Part Start Amendment Part6. The authority citation for part 347 continues to read as follows:
End Amendment Part Start Amendment Part7. Amend § 347.20 by lifting the stay on paragraph (e) (previously paragraph (d), redesignated at 74 FR 9765, March 6, 2009) and revising paragraph (e) to read as follows:
End Amendment PartStart Amendment PartPermitted combinations of active ingredients.* * * * *(e) Combinations of skin protectant and sunscreen active ingredients. Any one (two when required to be in combination) or more of the skin protectant active ingredients identified in § 347.10(a), (d), (e), (g), (h), (i), (k), (l), (m), and (r) of this chapter may be combined with any single sunscreen active ingredient identified in § 352.10 of this chapter, or any permitted combination of these ingredients identified in § 352.20 of this chapter, provided the product meets the conditions in § 352.20(b) of this chapter and is labeled according to §§ 347.60 and 352.60 of this chapter.
8. Amend § 347.60 by revising paragraphs (a), (b)(3), (c)(1), and (d)(1) to read as follows:
End Amendment PartStart PartLabeling of permitted combinations of active ingredients.* * * * *(a) Statement of identity. (1) Except as set forth in paragraph (a)(3) of this section, for a combination drug product that has an established name, the labeling of the product states the established name of the combination drug product, followed by the statement of identity for each ingredient in the combination, as established in the statement of identity sections of the applicable OTC drug monographs.
(2) Except as set forth in paragraph (a)(3) of this section, for a combination drug product that does not have an established name, the labeling of the product states the statement of identity for each ingredient in the combination, as established in the statement of identity sections of the applicable OTC drug monographs.
(3) For a product containing a combination of skin protectant and sunscreen active ingredients, the labeling of the product bears the statement of identity set forth in § 352.60(a) of this chapter.
(b) * * *
(3) Combinations of skin protectant and sunscreen active ingredients in § 347.20(e). In addition to any or all of the indications for skin protectant drug products in § 347.50(b)(2)(i) of this chapter, the required indications for sunscreen drug products in § 352.60(b) of this chapter must be used and any or all of the additional indications for sunscreen drug products may be used.
(c) * * *
(1) For combinations containing a skin protectant and a sunscreen identified in §§ 347.20(e) and 352.20(b). The warnings in § 352.60(c) of this chapter are used.
* * * * *(d) * * *
(1) For combinations containing a skin protectant and a sunscreen identified in §§ 347.20(e) and 352.20(b). The directions in § 352.60(d) of this chapter are used.
* * * * *PART 352—SUNSCREEN DRUG PRODUCTS FOR OVER-THE-COUNTER HUMAN USE
End Part Start Amendment Part9. The authority citation for part 352 continues to read as follows:
End Amendment Part Start Amendment Part10. Lift the stay of 21 CFR part 352.
End Amendment PartStart Amendment Part[Amended]11. In paragraph (a), remove the words “in a form suitable for topical administration”.
End Amendment Part Start Amendment Part12. Revise § 352.3 to read as follows:
End Amendment PartStart Amendment PartDefinitions.As used in this part:
(a) [Reserved]
(b) [Reserved]
(c) Sunscreen active ingredient. An active ingredient listed in § 352.10 that absorbs, reflects, or scatters radiation in the ultraviolet (UV) range at wavelengths from 290 to 400 nanometers.
(d) Determined sun protection factor (SPF) value. The SPF value that equals the largest whole number less than SPF − (t * SE), determined for a sunscreen product in accordance with § 201.327(i) of this chapter.
(e) Labeled sun protection factor (SPF) value. The SPF value associated with the range into which the determined SPF value falls, as set forth in the table in § 201.327(b)(2)(i) of this chapter.
(f) Extremely flammable. The term “extremely flammable” applies to any product that has a flashpoint at or below 20 °F (−6.7 °C) as determined by the test method described at 16 CFR 1500.43a, except that any product having one component or more with a flashpoint higher than 20 °F (−6.7 °C) that comprises at least 99 percent of the total volume of the product is not considered to be extremely flammable.
(g) Flammable. The term “flammable” applies to any product that has a flashpoint above 20 °F (−6.7 °C) and below 100 °F (37.8 °C) as determined by the test method described at 16 CFR 1500.43a, except that:
(1) Any product having one component or more with a flashpoint at or above 100 °F (37.8 °C) that comprises at least 99 percent of the total volume of the product is not considered to be flammable; and
(2) Any product containing 24 percent or less of water miscible alcohols, by volume, in aqueous solution is not considered to be flammable if the product does not present a significant flammability hazard when used by consumers.
(h) Combustible. The term “combustible” applies to any product having a flashpoint at or above 100 °F (37.8 °C) to and including 150 °F (65.6 °C) as determined by the test method described at 16 CFR 1500.43a, except that:
(1) Any product having one component or more with a flashpoint higher than 150 °F (65.6 °C) that comprises at least 99 percent of the total volume of the product is not considered to be combustible; and
(2) Any product containing 24 percent or less of water miscible alcohols, by volume, in aqueous solution is not considered to be combustible if the product does not present a significant flammability hazard when used by consumers.
13. Add § 352.5 to subpart A to read as follows:
End Amendment PartStart Amendment PartSun protection factor related conditions.(a) The product has a determined SPF value of at least 2 but no greater than 80.
(b) If the product has a determined SPF value of at least 15, it also passes Start Printed Page 6274the broad spectrum test in § 201.327(j) of this chapter.
14. Revise § 352.10 to read as follows:
End Amendment PartStart Amendment PartSunscreen active ingredients.The active ingredient of the product consists of any of the following, under the conditions specified, including being within the concentration specified for each ingredient:
(a) through (o) [Reserved]
(p) Titanium dioxide up to 25 percent
(q) [Reserved]
(r) Zinc oxide up to 25 percent.
15. Revise § 352.20 to read as follows:
End Amendment PartStart Amendment PartPermitted combinations of active ingredients.The determined SPF of any product containing a sunscreen active ingredient is measured by the testing procedures established in § 201.327(i) of this chapter.
(a) Combinations of sunscreen active ingredients. Two or more sunscreen active ingredients identified in § 352.10 may be combined with each other in a single sunscreen product if all of the following conditions are met:
(1) Each sunscreen active ingredient in the product must satisfy the conditions established for its use in § 352.10.
(2) The concentration of each sunscreen active ingredient must be sufficient to contribute a minimum determined SPF of not less than 2 to the finished product.
(3) The finished product must have a minimum determined SPF of not less than the number of sunscreen active ingredients used in the product multiplied by 2.
(b) Combinations of sunscreen and skin protectant active ingredients. Any single sunscreen active ingredient identified in § 352.10 or any combination of sunscreen active ingredients permitted under paragraph (a) of this section may be combined with one or more skin protectant active ingredients identified in §§ 347.10(a), (d), (e), (g), (h), (i), (k), (l), (m), and (r) of this chapter when all of the following conditions are met:
(1) Each sunscreen active ingredient in the product must satisfy the conditions established for its use in § 352.10.
(2) The concentration of each sunscreen active ingredient must be sufficient to contribute a minimum determined SPF of not less than 2 to the finished product.
(3) The finished product must have a minimum determined SPF of not less than the number of sunscreen active ingredients used in the product multiplied by 2.
(4) The product must be labeled according to § 201.327(h) of this chapter and § 352.60.
(c) [Reserved]
16. Add § 352.30 to subpart B to read as follows:
End Amendment Part Start Amendment Part17. Add § 352.40 to subpart B to read as follows:
End Amendment PartStart Amendment PartDosage forms.The product is in one of the following dosage forms and meets any additional conditions specified:
(a) Oil.
(b) Lotion.
(c) Cream.
(d) Gel.
(e) Butter.
(f) Paste.
(g) Ointment.
(h) Stick.
(i) Spray, provided that all of the following conditions are satisfied:
(1) Size of particles as dispensed from the consumer container:
(i) No more than 10 percent of the particles dispensed from the consumer container are smaller than 10 micrometers; and
(ii) None of the particles dispensed from the consumer container are smaller than 5 micrometers.
(2) The product does not meet the definition of the term “extremely flammable” as defined in § 352.3(f).
(3) If the product meets the definition of either the term “flammable” or the term “combustible” as defined at §§ 352.3(g) or (h), as applicable, when tested in accordance with 16 CFR 1500.43a, the product also has a measured drying time of less than 10 minutes.
(4) The product is labeled as required by §§ 201.327(d) and (e)(5) of this chapter.
(5) Testing in accordance with part 211 of this chapter must confirm that the product meets the conditions for particle size, flammability, and drying time as required by this section and reflected in the product labeling.
(i) Testing of each lot of product for size of particles dispensed from the consumer container must be conducted in accordance with adequate written specifications.
(ii) Flammability testing for each batch of product must be conducted in accordance with the specifications set forth in 16 CFR 1500.43a.
(iii) If the product meets the definition of either the term “flammable” or “combustible” as defined at § 352(g) or (h), as applicable, when tested accordance with 16 CFR 1500.43a, drying time for each lot of product must be conducted in accordance with adequate written specifications.
18. Revise § 352.50 to read as follows:
End Amendment PartStart Amendment PartPrincipal display panel of all sunscreen drug products.The principal display panel labeling must comply with § 201.327(b) of this chapter.
19. Revise § 352.52 to read as follows:
End Amendment PartStart Amendment PartLabeling of products containing one or more sunscreen active ingredients.(a) Statement of identity. The labeling of the product contains the statement of identity, in accordance with § 201.327(b) of this chapter.
(b) Indications. The labeling of the product contains the indication statements identified in § 201.327(c) of this chapter, as appropriate, and subject to the conditions stated therein.
(c) Warnings. The labeling of the product contains the warnings in § 201.327(d) of this chapter, as applicable, under the heading “Warnings:”
(d) Directions. The labeling of the product contains the statements in § 201.327(e) of this chapter, as applicable, under the heading “Directions.”
(e) Other information. The labeling of the product contains the statement in § 201.327(f) of this chapter under the heading “Other information.”
(f) False or misleading claims. The labeling of the product must not contain any claims that would be false and/or misleading on sunscreen products, as outlined in § 201.327(g) of this chapter.
20. Revise § 352.60 to read as follows:
End Amendment PartStart Amendment PartLabeling of products containing a combination of sunscreen and skin protectant active ingredients.Statements of identity, indications, warnings, and directions for use, respectively, applicable to each ingredient in the product may be combined to eliminate duplicative words or phrases so that the resulting information is clear and understandable. Labeling provisions in § 347.50(e) of this chapter shall not apply to these products.
(a) Statement of identity. The labeling of the product bears the statement of identity, as set forth in § 201.327(h)(1) of this chapter.
(b) Indications. The labeling of the product states, under the heading “Uses,” the applicable indication statements, as set forth in § 201.327(h)(2) of this chapter.
(c) Warnings. The labeling of the product states, under the heading Start Printed Page 6275“Warnings,” the applicable warning statements, as set forth in § 201.327(h)(3) of this chapter.
(d) Directions. The labeling of the product states, under the heading “Directions,” the applicable direction statements, as set forth in § 201.327(h)(4) of this chapter.
21. Revise subpart D to read as follows:
End Amendment PartSubpart D—Final Formulation Testing
Start SignatureBroad spectrum testing.If the product's determined SPF value is at least 15, the product is tested and shown to pass the broad spectrum test in § 201.327(j) of this chapter.
End Signature End Supplemental InformationDated: February 14, 2019.
Scott Gottlieb,
Commissioner of Food and Drugs.
Footnotes
1. An OTC monograph establishes conditions under which certain OTC drugs may be marketed without approved new drug applications because they are generally recognized as safe and effective (GRASE) and not misbranded. The proposed rule classifies active ingredients and other conditions as Category I (proposed to be GRASE and not misbranded), Category II (proposed to be not GRASE or to be misbranded), or Category III (additional data needed).
Back to Citation2. Unless otherwise noted, references in this proposed rule to sunscreen active ingredients and/or sunscreen products are to sunscreen active ingredients or products marketed pursuant to the OTC monograph system and subject to 21 CFR 201.327. Unless specifically noted, references to sunscreen active ingredients and/or sunscreen products in this notice do not refer to those marketed pursuant to a new drug application (NDA) or an abbreviated new drug application (ANDA). They also do not refer to sunscreen active ingredients being evaluated under the new procedures set out in the SIA (21 U.S.C. 360fff et seq).
Back to Citation3. We note that because our proposal to raise the maximum labeled SPF value to 60+ is based on studies that all used broad spectrum sunscreens, the additional clinical benefit we are proposing to recognize in sunscreen products with SPF values greater than 50 cannot be decoupled from the broad spectrum protection provided by those products. As a result, our proposal to raise the maximum labeled SPF value to SPF 60+ is both consistent with and dependent upon our proposal to require that all sunscreen monograph products with SPF values of 15 and above satisfy our broad spectrum requirements.
Back to Citation4. We note that, for ease of comprehension, we have included in this document the current provisions of 21 CFR 201.327 that we are not proposing to revise along with the provisions of that regulation that we are proposing to revise.
Back to Citation5. As described in further detail in section IXB.2, in the time since the L&E Final Rule was issued in 2011, the body of evidence about the role of UVA radiation in the development of skin cancer has grown. As a result, FDA is making a number of proposals designed (among other things) to couple a greater magnitude of UVA protection to increases in SPF values.
Back to Citation6. The ingredients were: Aminobenzoic acid, digalloyl trioleate, 2-ethylhexyl 2-cyano-3,3-diphenylacrylate, glyceryl aminobenzoate, menthyl anthranilate, padimate O, sulisobenzone, cinoxate, dioxybenzone, ethylhexyl p-methoxycinnamate, homosalate, oxybenzone, 2-phenylbenzimidazole-5-sulfonic acid, titanium dioxide, diethanoloamine p-methoxycinnamate, ethyl 4-[bis (hydroxylpropyl)] aminobenzoate, 2-ethylhexyl salicylate, lawsone with dihydroxyacetone, padimate A, red petrolatum, and triethanolamine salicylate.
Back to Citation7. In 61 FR 48645 (September 16, 1996) (proposing that avobenzone is GRASE up to 3 percent alone and 2 to 3 percent when in combination with cinoxate, diethanolamine methoxycinnamate, dioxybenzone, homosolate, octocrylene, octyl methoxycinnamate, octyl salicylate, oxybenzone, sulisobenzone, and/or trolamine salicylate) and 63 FR 56584 (October 22, 1998) (proposing that zinc oxide is GRASE alone or in combination with any previously proposed GRASE active ingredient except avobenzone). The list of active ingredients was (and would continue to be) modified because of, among other things, a lack of interest in developing United States Pharmacopeia (USP) compendial monographs for certain of the active ingredients originally proposed (see 64 FR 27666 at 27681).
Back to Citation8. See § 352.10, now stayed; 64 FR 27666. The active ingredient names used in that regulation, as originally published, differ from those used in table 1, which are the current established names for these active ingredients. We note that subsequent to the publication of the Stayed 1999 Final Monograph, we issued another final rule in 2002 amending the names used for four of those ingredients to make them consistent with the renaming of those ingredients in the corresponding USP monographs (67 FR 41821 at 41823, June 20, 2002). Under section 502(e) of the FD&C Act, drug labels are required to bear the established name of each active ingredient, and if FDA has not designated an official name under section 508 of the FD&C Act (21 U.S.C. 358), the compendial name is the established name. To comply with section 502(e) of the FD&C Act, sunscreen drug products must therefore bear the current compendial names for their active ingredients, and the current compendial names are used throughout this document. Because the 2002 final rule that changed those names was published after part 352 was stayed, however, those amendments have not yet been incorporated into the published monograph regulation.
Back to Citation9. An exception to this rule involving avobenzone was retained from the TFM: The Stayed 1999 Final Monograph stated that avobenzone may not be combined with PABA, phenylbenzimidazole sulfonic acid, menthyl anthranilate, padimate O, titanium dioxide, or zinc oxide. In 2007, we proposed to include in the monograph a condition permitting the marketing of sunscreens containing avobenzone in combination with either zinc oxide or ensulizole based on safety and effectiveness data about these combinations provided to the docket (“Sunscreen Drug Products for Over-the-Counter Human Use: Proposed Amendment of Final Monograph”, 72 FR 49070 at 49074, August 27, 2007). As described in section VII.A, we now anticipate finalizing a monograph that would permit all listed active ingredients to be combined without limitation. This approach is consistent with the approach to sunscreen combinations generally taken throughout the OTC Drug Review for sunscreens.
Back to Citation10. FDA's proposed sunscreen orders on each of these ingredients can be found at https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/ucm434843.htm.
Back to Citation11. FDA's recommendations regarding the safety and effectiveness data necessary to determine whether an OTC sunscreen active ingredient (or combination of ingredients) evaluated under the SIA was GRASE when used under specified conditions generally remained unchanged in the final guidance.
Back to Citation12. Chronic use is defined as continuous or intermittent use for at least 6 months during the course of a lifetime.
Back to Citation13. Cmax is the peak plasma concentration and Tmax is the time to peak plasma concentration.
Back to Citation14. We note, however, as described in section VIII.C.1.b, that because of avobenzone's potential for photodegradation, we recommend that a MUsT for avobenzone evaluate avobenzone in combination with a photostabilizer. In some cases, sunscreen active ingredients (e.g., octocrylene) can serve as photostabilizers. In such cases, we expect that the MUsT could include such ingredients.
Back to Citation15. FDA has issued draft guidance with recommendations for the conduct of MUsT studies to support the safety of active ingredients that are candidates for inclusion in a topical drug product under an OTC Drug monograph (Ref. 16). When finalized, this guidance will represent FDA's current thinking on this topic. FDA also encourages persons who are interested in conducting a MUsT to support the safety of an active ingredient to discuss proposed protocols with the Agency.
Back to Citation16. As discussed infra, the MUsT should be conducted on healthy, intact skin because sunscreens are intended for prevention rather than treatment.
Back to Citation17. FDA recommends submitting the carcinogenicity study protocol(s) for review by FDA's Center for Drug Evaluation and Research's (CDER's) Executive Carcinogenicity Assessment Committee before initiating the studies. For further guidance regarding carcinogenicity studies, see the FDA guidance for industry “Carcinogenicity Study Protocol Submissions,” May 2002 (available at https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm078924.pdf).
Back to Citation18. Examples of such pathways could include endocrine function and signaling pathways related to growth and development.
Back to Citation19. We note that nanoscale zinc oxide can be solubilized to a small extent in the presence of phosphate and lecithin at pH's that are achievable on the skin. Even under these conditions, however, the amount potentially absorbed is de minimis and far lower than daily nutritional intake of zinc.
Back to Citation20. This literature included three clinical safety studies conducted by Hill Top Research, Inc. for Procter & Gamble regarding (a) human sensitization (Study Reports 96-6635-76a and 96-6635-76b); (b) human photoirritation/phototoxicity (Study Report 96-6634-76); and (c) human photoallergenicity (Study Report 96-6633-76). See Citizen Petition submitted by Proctor & Gamble, June 24, 1997 (FDA-1978-N-0018-0639) and the “Opinion concerning Zinc Oxide” drafted by the European Commission, Scientific Committee on Cosmetic Products and Non-Food Products Intended for Consumers (SCCNFP), which included five summaries of human clinical safety studies, all evaluating zinc oxide 25 percent (Ref. 40).
Back to Citation21. See, e.g., Beeckman et al. (Ref. 41); 43 FR 34628 at 34641(August 4, 1978) (discussing use of zinc oxide 1 percent to 25 percent as a skin protectant active ingredient: “Zinc oxide is widely recognized as a skin protectant” and “No reports of topical toxicity were found in the literature” on zinc oxide).
Back to Citation22. Our review of the available nonclinical safety literature on zinc oxide included references for a 90-day dermal toxicity study, genotoxicity, and limited developmental and reproductive toxicity information. The review of this literature suggests that genotoxicity, findings for zinc oxide are mixed, and that there is minimal dermal toxicity in rodents after 90 days. (See Refs. 42 and 43.) Oral rat embryofetal toxicity studies showed some adverse maternal and fetal effects, but only at very high doses (>200 mg/kg/day) significantly higher than what is at issue here (Refs. 44 and 45).
Back to Citation23. We note that the available literature also includes data showing that oral administration of relatively high doses of titanium dioxide did not produce adverse fetal effects in rats. (See Ref. 56.)
Back to Citation24. In a July 2013 opinion addressing the safe use of titanium dioxide in sunscreen products, the European Commission's Scientific Committee on Consumer Safety gave its opinion that titanium dioxide particles consisting, among other things, of up to 5 percent anatase crystal “can be considered to not pose any risk of adverse effects in humans after application on healthy, intact or sunburnt skin” (Ref. 62). In 2016, this physicochemical parameter was incorporated by the European Commission into its Regulation on Cosmetic Products (Regulation (EC) No. 1223/2009 11/30/2009) permitting the use of titanium dioxide as a UV filter or as a colorant in cosmetics. See Regulation (EC) No 1143/2016 July 13, 2016.
Back to Citation25. Id.
Back to Citation26. Total sunscreen sales since 1969 are not readily available. However, in 2016 a total of 161,882,779 sunscreen units were sold in the United States (Ref. 68).
Back to Citation27. This direction applies to sunscreens with an SPF of 15 or greater that are also broad spectrum.
Back to Citation28. Based on an evaluation of product labeling available at https://labels.fda.gov (accessed April 4, 2018). See also External Analgesic TFM.
Back to Citation29. The symptoms associated with both acute and chronic salicylate toxicity are well established. Descriptions are available from many sources, including: National Library of Medicine's Toxicology Data Network (ToxNet), “Salicylic Acid,” September 2008, available at https://toxnet.nlm.nih.gov/cgi-bin/sis/search/a?dbs+hsdb:@term+@DOCNO+672 (accessed March 27, 2018). FDA also included a comprehensive summary of salicylism in 21 CFR 343.80.
Back to Citation30. In mice, liver tumors were identified, providing evidence of systemic absorption of trolamine, but the suspected mechanism of action is likely not relevant to humans (Refs. 79 and 80). A causal link between the proposed mechanism and tumor formation in mice is lacking.
Back to Citation31. These changes could potentially be addressed with historical control data (Ref. 88).
Back to Citation32. For examples of the type of studies that could be explored at that juncture see Ref. 21.
Back to Citation33. As a reminder, such data must be generally available to be considered as part of this rulemaking process. Once available, FDA intends to review such data to determine whether it resolves particular data concerns we have in this area.
Back to Citation34. Reactive oxygen species are “a type of unstable molecule that contains oxygen and that easily reacts with other molecules in a cell. A buildup of ROS in cells may cause damage to DNA, RNA, and proteins, and may cause cell death.” https://www.cancer.gov/publications/dictionaries/cancer-terms?cdrid=687227.
Back to Citation35. Avobenzone's photodegradation also results in the formation of free radicals, which could, in theory, create sensitization and irritation responses and increase long-term risk of skin cancers and photoaging (Ref. 139).
Back to Citation36. FDA-1978-N-0018-0675, two volume submission (February 20, 2009) (L'Oreal Petition).
Back to Citation37. Id., volume I, pp. 5-8.
Back to Citation38. The available nonclinical data for avobenzone include acute oral and dermal toxicity studies in rats; a 13-week oral toxicity study in rats; a 28-day dermal toxicity study in rats; a 21-day dermal toxicity study in rabbits; several in vitro genotoxicity tests; an in vivo micronucleus test in mice, as well as a sensitization test in guinea pigs; a primary skin irritation test in rabbits; an ocular irritation test in rabbits; a phototoxicity study in guinea pigs; a photoallergenicity study in guinea pigs; and embryofetal development studies in rats and rabbits (Givaudan-Roure Petition, Docket No. FDA-1978-N-0018-0751). Importantly, (except for the embryofetal development studies) these studies are not sufficient to resolve safety concerns for a chronically used product.
Back to Citation39. See § 201.327 for the current labeling requirements, and underlying testing, for OTC sunscreens containing one or more of the 16 active ingredients that are addressed in this rulemaking, for use in products marketed without approved NDAs. OTC sunscreens marketed under NDAs provide similar information in their product-specific applications to substantiate their labeling. For proposed changes to § 201.327, see codified section of this document. The Stayed 1999 Final Monograph also required SPF testing of final formulations as a GRASE condition. Elsewhere in this proposed rule, we propose to establish monograph conditions in 21 CFR part 352 that ensure that all sunscreens are tested for SPF in accordance with § 201.327(i) and achieve a minimum SPF of 2, and that certain sunscreens pass the broad spectrum test in § 201.327(j).
Back to Citation40. We note that, as described in section IX.D.2.i, we are proposing a minor revision in equipment specifications for the broad spectrum test to respond to feedback that FDA received on this issue and proposing some minor revisions to current language to make clear our existing expectations.
Back to Citation41. As noted in section III.A.2, only those broad spectrum sunscreen products that have an SPF of 15 or higher have been shown to help prevent skin cancer and early skin aging.
Back to Citation42. As used in this preamble, the determined SPF value is the SPF value that equals the largest whole number less than SPF−(t*SE), determined for a sunscreen product in accordance with § 201.327(i). See also section IX.D.2.b, where we propose to define this term in the regulation.
Back to Citation43. The minimal erythema dose (MED) is the smallest UV dose that produces perceptible redness of the skin (erythema) with clearly defined borders at 16 to 24 hours after UV exposure (§ 201.327(i)(5)(i)).
Back to Citation44. The determination of SPF for each subject is calculated via a ratio of the MED of protected skin over the final MED of unprotected skin. In a scenario in which the final MED of unprotected skin is underestimated by 15 percent and the MED of protected skin is overestimated by 15 percent, this would present approximately 30 percent variability for the individual subject.
Back to Citation45. We note that the use of ranges to represent SPF values on product labeling is already in use in Australia and the European Union (Refs. 180 and 181).
Back to Citation46. The proposed labeled values are expressed in increments of 5 for products with determined SPF results of 15 to 29.9 (i.e., SPF 15, SPF 20, SPF 25), but for determined SPF results of 30 or more, the proposed labeled values are expressed in increments of 10 (i.e., SPF 30, SPF 40, SPF 50, with a proposed maximum of SPF 60+.).
Back to Citation47. EPA Product List (Ref. 187); a similar list of insecticide products on the National Pesticide Information Center (NPIC) website produced similar results (Ref. 188).
Back to Citation48. Some insect repellents are also regulated by FDA as human drugs (e.g., pediculicides and scabicides intended to control parasites on humans) or animal drugs (e.g., pesticide products for oral administration to animals) (7 U.S.C. 136 et seq.); see also “MOU 225-73-8010 Memorandum of Understanding Between the Environmental Protection Agency and the United States Department of Health, Education and Welfare Food and Drug Administration,” (available at https://www.fda.gov/aboutfda/partnershipscollaborations/memorandaofunderstandingmous/domesticmous/ucm115873.htm (accessed April 17, 2018).
Back to Citation49. DEET 2014 reregistration interim review final decision (EPA-HQ-OPP-2012-0162) (available at https://www3.epa.gov/pesticides/chem_search/reg_actions/reregistration/red_PC-080301_1-Apr-98.pdf (accessed April 17, 2018).
Back to Citation50. Id. In June 2014, EPA issued an interim review of DEET and did not identify any specific new concerns. The proposed interim registration review decision became final on September 24, 2014. DEET 2014 reregistration interim review final decision (EPA-HQ-OPP-2012-0162) (available at https://www.regulations.gov/document?D=EPA-HQ-OPP-2012-0162-0012 (accessed April 17, 2018).
Back to Citation51. DEET 2014 reregistration interim review final decision, supra note 49, v.
Back to Citation52. The primary estimate of the costs is not the average of the lower bound costs and the upper bound costs.
Back to CitationBILLING CODE 4164-01-P
BILLING CODE 4164-01-C
BILLING CODE 4164-01-C
[FR Doc. 2019-03019 Filed 2-21-19; 8:45 am]
BILLING CODE 4164-01-P
















Document Information
- Published:
- 02/26/2019
- Department:
- Food and Drug Administration
- Entry Type:
- Proposed Rule
- Action:
- Proposed rule.
- Document Number:
- 2019-03019
- Dates:
- Submit either electronic or written comments. on the proposed rule by May 28, 2019. Electronic comments must be submitted on or before May 28, 2019. The https://www.regulations.gov electronic filing system will accept comments until 11:59 p.m. Eastern Time at the end of May 28, 2019. See section XII for proposed effective and compliance dates of a final rule based on this document.
- Pages:
- 6204-6275 (72 pages)
- Docket Numbers:
- Docket No. FDA-1978-N-0018
- RINs:
- 0910-AF43: Sunscreen Drug Products For Over-The-Counter-Human Use; Final Monograph
- RIN Links:
- https://www.federalregister.gov/regulations/0910-AF43/sunscreen-drug-products-for-over-the-counter-human-use-final-monograph-
- Topics:
- Administrative practice and procedure, Drugs, Incorporation by reference, Labeling, Medical devices, Over-the-counter drugs, Reporting and recordkeeping requirements
- PDF File:
- 2019-03019.pdf
- CFR: (17)
- 21 CFR 201.327
- 21 CFR 310.545
- 21 CFR 310.549
- 21 CFR 347.20
- 21 CFR 347.60
- More ...