07-4147. Two Optional Methods for Relative Accuracy Test Audits of Mercury Monitoring Systems Installed on Combustion Flue Gas Streams and Several Amendments to Related Mercury Monitoring Provisions
-
Start Preamble
Start Printed Page 51494
AGENCY:
Environmental Protection Agency (EPA).
ACTION:
Direct final rule.
SUMMARY:
EPA is taking direct final action on two optional methods for relative accuracy audits of mercury monitoring systems installed on combustion flue gas streams and several amendments to related mercury monitoring provisions. This action approves two optional mercury (Hg) emissions test methods for potential use in conjunction with an existing regulatory requirement for Hg emissions monitoring, as well as several revisions to the mercury monitoring provisions themselves. This action is in regard to the testing and monitoring requirements for mercury specified in the Federal Register on May 18, 2005. Since that publication, EPA has received numerous comments concerning the desirability of EPA evaluating and allowing use of the measurement techniques addressed in the two optional methods in lieu of the methods identified in the cited Federal Register publication, as they can produce equally acceptable measures of the relative accuracy achieved by Hg monitoring systems. This action allows use of these two optional methods entirely at the discretion of the owner or operator of an affected emission source in place of the two currently specified methods. This direct final rule also amends Performance Specification 12A by adding Methods 30A and 30B to the list of reference methods acceptable for measuring Hg concentration and the Hg monitoring provisions of May 18, 2005, to reflect technical insights since gained by EPA which will help to facilitate implementation including clarification and increased regulatory flexibility for affected sources.
DATES:
This rule is effective on November 6, 2007 without further notice, unless EPA receives adverse comment by October 9, 2007. If EPA receives adverse comment, EPA will publish a timely withdrawal in the Federal Register informing the public that some or all of the amendments in this rule will not take effect.
ADDRESSES:
Submit your comments, identified by Docket ID No. EPA-HQ-OAR-2007-0164, by one of the following methods:
- www.regulations.gov. Follow the on-line instructions for submitting comments.
- E-mail: a-and-r-docket@epa.gov.
- Fax: (202) 566-9744.
- Mail: Two Optional Methods for Relative Accuracy Test Audits of Mercury Monitoring Systems Installed on Combustion Flue Gas Streams and Several Amendments to the Related Mercury Monitoring Provisions, Environmental Protection Agency, Mailcode: 2822T, 1200 Pennsylvania Avenue, NW., Washington, DC 20460. Please include a total of two copies.
- Hand Delivery: EPA Docket Center, 1301 Constitution Avenue, NW., EPA Headquarters Library, Room 3334, EPA West Building, Washington, DC 20460. Such deliveries are only accepted during the Docket's normal hours of operation, and special arrangements should be made for deliveries of boxed information.
Instructions: Direct your comments to Docket ID No. EPA-HQ-OAR-2007-0164. EPA's policy is that all comments received will be included in the public docket without change and may be made available online at www.regulations.gov, including any personal information provided, unless the comment includes information claimed to be Confidential Business Information (CBI) or other information whose disclosure is restricted by statute. Do not submit information that you consider to be CBI or otherwise protected through www.regulations.gov or e-mail. The www.regulations.gov Web site is an “anonymous access” system, which means EPA will not know your identity or contact information unless you provide it in the body of your comment. If you send an e-mail comment directly to EPA without going through www.regulations.gov, your e-mail address will be automatically captured and included as part of the comment that is placed in the public docket and made available on the Internet. If you submit an electronic comment, EPA recommends that you include your name and other contact information in the body of your comment and with any disk or CD-ROM you submit. If EPA cannot read your comment due to technical difficulties and cannot contact you for clarification, EPA may not be able to consider your comment. Electronic files should avoid the use of special characters, any form of encryption, and be free of any defects or viruses. For additional information about EPA's public docket, visit the EPA Docket Center homepage at http://www.epa.gov/epahome/dockets.htm.
Docket: All documents in the docket are listed in the www.regulations.gov index. Although listed in the index, some information is not publicly available, e.g., CBI or other information whose disclosure is restricted by statute. Certain other material, such as copyrighted material, will be publicly available only in hard copy. Publicly available docket materials are available either electronically in www.regulations.gov or in hard copy at the Two Optional Methods for Relative Accuracy Audits of Mercury Monitoring Systems Installed on Combustion Flue Gas Streams Air and Radiation Docket, EPA/DC, EPA West Building, EPA Headquaters Library, Room 3334, 1301 Constitution Avenue, NW., Washington, DC. The Public Reading Room is open from 8:30 a.m. to 4:30 p.m., Monday through Friday, excluding legal holidays. The telephone number for the Public Reading Room is (202) 566-1744, and the telephone number for the Air and Radiation Docket is (202) 566-1742.
Start Further InfoFOR FURTHER INFORMATION CONTACT:
Either Mr. William Grimley, Office of Air Quality Planning and Standards, Air Quality Assessment Division, Measurement Technology Group (E143-02), EPA, Research Triangle Park, NC 27711, telephone (919) 541-1065, facsimile number (919) 541-0516, e-mail address: grimley.william@epa.gov or Ms. Robin Segall, Office of Air Quality Planning and Standards, Air Quality Assessment Division, Measurement Technology Group (E143-02), EPA, Research Triangle Park, NC 27711, telephone (919) 541-0893, facsimile number (919) 541-0516, e-mail address: segall.robin@epa.gov.
End Further Info End Preamble Start Supplemental InformationSUPPLEMENTARY INFORMATION:
I. Why is EPA using a Direct Final Rule?
EPA is publishing this rule without a prior proposed rule because we view this as a noncontroversial action and anticipate no adverse comment. The most important benefit of direct final rulemaking for this action is to provide: (1) Additional reference method options, and (2) judicious revisions to mercury monitoring provisions specified in the Federal Register on May 18, 2005 that, if successful, relieve affected facilities of uncertainty regarding final emission monitoring requirements and certification details as opposed to waiting through a potentially protracted proposal/final Start Printed Page 51495rulemaking process. Insofar as the two methods are concerned, EPA believes that they contain the necessary elements to generate acceptable data quality without being unduly burdensome. Through experience gained from developing existing performance based methods and trading rules, EPA has learned to identify test method criteria significant to effective rule implementation. EPA believes each of the two methods adopted in this action contain adequate specific criteria and procedures essential to the accurate measurement of Hg emissions, without adversely compromising the goals of performance-based methodology. EPA will continue to support and advance the principles and practicality of these methods by adding detailed method application information to facilitate their use to the Web site www.epa.gov/airmarkets/ as it becomes available. Since use of either of these methods is not mandatory, but optional, there should be no objection to their availability. Regarding the amendments to the Hg emission monitoring provisions of 40 CFR parts 72 and 75, these amendments reflect EPA's increased technical understanding since the May 18, 2005 rulemaking. However, in the “Proposed Rules” section of today's Federal Register, we are publishing a separate document that will serve as the proposed rule to approve provisions, if any, of this direct final rule that receive relevant adverse comments on this direct final rule. We will not institute a second comment period on this action. Any parties interested in commenting must do so at this time. For further information about commenting on this rule, see the ADDRESSES section of this document.
If EPA receives adverse comment on one or more distinct provisions of this rulemaking, we will publish a timely withdrawal in the Federal Register indicating which provisions we are withdrawing and informing the public that those provisions will not take effect. The provisions that are not withdrawn will become effective on the date set out above, notwithstanding adverse comment on any other provision. We would address all public comments in a subsequent final rule based on the proposed rule.
II. Does This Action Apply to Me?
Regulated Entities. The regulated categories and entities affected by this direct final rule include:
Category NAICS a Examples of regulated entities Industry 221112 Fossil fuel-fired electric utility steam generating units. Federal government b 221122 Fossil fuel-fired electric utility steam generating units owned by the Federal government. State/local governments b 221122 Fossil fuel-fired electric utility steam generating units owned by municipalities. Tribal governments 921150 Fossil fuel-fired electric utility steam generating units in Indian country. a North American Industry Classification System. b Federal, State, or local government-owned and operated establishments are classified according to the activity in which they are engaged. This table is not intended to be exhaustive, but rather provides a guide for readers regarding entities likely to be affected by this direct final rule. If you have any questions regarding the applicability of this direct final rule to a particular entity, consult either the air permit authority for the entity or your EPA regional representative as listed in 40 CFR 63.13.
III. Where Can I Obtain a Copy of This Action?
In addition to being available in the docket, an electronic copy of this direct final rule is also available on the World Wide Web through the Technology Transfer Network (TTN). Following signature, a copy of this direct final rule will be posted on the TTN's policy and guidance page for newly proposed or promulgated rules at the following address: http://www.epa.gov/ttn/oarpg. The TTN provides information and technology exchange in various areas of air pollution control.
IV. How Is This Document Organized?
The information presented in this preamble is organized as follows:
I. Why Is EPA Using a Direct Final Rule?
II. Does This Action Apply to Me?
III. Where Can I Obtain a Copy of This Action?
IV. How Is This Document Organized?
V. Background
VI. This Action
VII. Statutory and Executive Order Reviews
A. Executive Order 12866: Regulatory Planning and Review
B. Paperwork Reduction Act
C. Regulatory Flexibility Act
D. Unfunded Mandates Reform Act
E. Executive Order: 13132: Federalism
F. Executive Order 13175: Consultation and Coordination With Indian Tribal Governments
G. Executive Order 13045: Protection of Children From Environmental Health and Safety Risks
H. Executive Order 13211: Actions That Significantly Affect Energy Supply, Distribution, or Use
I. National Technology Transfer and Advancement Act
J. Executive Order 12898: Federal Actions To Address Environmental Justice in Minority Populations and Low-Income Populations
K. Congressional Review Act
V. Background
On May 18, 2005, in the preamble of the Clean Air Mercury Rule (CAMR) (70 FR 28608), EPA stated its intention to propose and promulgate an instrumental reference method as an alternative to the use of ASTM Method D6784-02 (the Ontario Hydro Method) to perform Relative Accuracy Test Audits (RATAs) of Hg continuous emission monitoring systems (CEMS) and sorbent trap monitoring systems used to monitor Hg emissions from coal-fired power plants.
In comments on the proposed CAMR, commenters had two primary objections to the use of the Ontario Hydro Method as the reference test method for RATAs. Some expressed concern that the complexity of this wet chemical method could lead to results that would cause a properly functioning Hg CEMS to fail a RATA. Other commenters noted that, unlike instrumental reference methods used to audit CEMS for SO2 and NOX that provide real-time values, test results from the Ontario Hydro Method can take weeks to be received from the laboratory. Commenters stated that this time lag can lead to implementation problems with regard to both missing data and emissions reporting.
Since the CAMR was promulgated, EPA has proposed changes to 40 CFR part 75, which would allow the use of EPA Method 29, with enhanced quality-assurance procedures, as an alternative Hg reference method (71 FR 49257; August 22, 2006). Although Method 29 is somewhat simpler than the Ontario Hydro Method and is more familiar to stack testers and State regulatory agencies, it is also a wet chemistry method and is, therefore, subject to the same limitations that make the Ontario Hydro method less than optimal for RATA testing.
In view of these considerations, EPA believes that for RATA testing, an instrumental Hg reference method Start Printed Page 51496would be preferable to both the Ontario Hydro Method and to Method 29. An instrumental method would provide real-time data that would best facilitate implementation of a mercury cap and trade program. Therefore, this action approves a performance-based instrumental reference method for measuring Hg emission concentrations.
Another commenter to the proposed CAMR recommended that the sorbent trap monitoring approach, now specified in 40 CFR part 75, appendix K, be considered for use as a reference method. Although EPA did not commit to establishing a sorbent trap reference method at the time of CAMR promulgation, stakeholder interest in this methodology has increased significantly. In an August 22, 2006 Federal Register notice, EPA solicited comment on the use of sorbent trap technology for Hg reference method testing, and numerous supportive comments were received. In view of this, we initiated a review of available historical test data where concurrent measurements of Hg concentration were made with sorbent trap systems and either the Ontario Hydro Method or Method 29. These data, taken together with additional supporting data from recent field tests that were performed after the CAMR was promulgated, suggest that using the sorbent trap methodology for Hg reference method testing is viable. The Hg sorbent trap approach is less onerous to use than either Ontario Hydro or Method 29, and although it does not measure real-time Hg concentrations, a thermal technique can be used to analyze the samples on the same day that they are collected, facilitating RATA testing in the context of a cap and trade program. Therefore, this action also approves a sorbent trap reference method for Hg, as an alternative to the Ontario Hydro Method and Method 29.
This direct final rule also includes several carefully considered amendments to the Hg emission monitoring provisions of 40 CFR parts 72 and 75. EPA believes these amendments will facilitate implementation of the CAMR by clarifying portions of that rule and by providing added regulatory flexibility to the affected sources.
VI. This Action
This direct final rule allows for the earliest possible use of two optional reference test methods for measuring total vapor phase mercury emissions from stationary sources as well as several related amendments to the Hg monitoring provisions of the CAMR. Both an instrumental test method and a sorbent trap test method for measurement of total vapor phase mercury emissions are being added to Appendix A-8 of 40 CFR part 60 as approved alternatives to the Ontario Hydro Method and EPA Method 29 to perform RATAs of installed mercury monitoring systems. The two methods are discussed below, and the related amendments are explained in detail later in this section.
The first method being added to appendix A-8 of 40 CFR part 60 today is titled “Method 30A—Determination of Total Vapor Phase Mercury Emissions from Stationary Sources (Instrumental Analyzer Procedure).” In Method 30A, a sample of the effluent gas is continuously extracted and conveyed to an analyzer capable of measuring the total vapor phase Hg concentration. Elemental and oxidized mercury (i.e., Hg0 and Hg+2) may be measured separately or simultaneously but, for purposes of this method, total vapor phase Hg is the sum of Hg0 and Hg+2. Method 30A provides test program-specific verification of method performance using a dynamic spiking approach, coupled with other performance criteria, which include system calibration, interference testing, and system integrity/drift checks. The dynamic spiking requirement, which is a gaseous “method of standard additions,” is the only part of Method 30A not parallel to the routinely applied instrumental reference methods used to perform relative accuracy testing of CEMS for SO2 and NOX. The dynamic spiking procedure is included in Method 30A to characterize measurement bias for Hg, which can be highly reactive on a site-specific basis (i.e., for each emissions sample matrix), with recovery criteria set to ensure that the bias is held to a minimal level. All performance requirements of Method 30A must be met for the data to be considered valid. The availability of an instrumental reference method for Hg testing is consistent with the approach EPA has taken in the successful Acid Rain and NOX Budget emissions trading programs.
Method 30A is performance based in keeping with the criteria established under our Notice of Intent to Implement Performance Based Measurement Systems for Environmental Monitoring (62 FR 52098, October 6, 1997). Use of the performance-based measurement approach will allow for continued development and application of new, improved, and more cost-effective Hg measurement technologies while ensuring the collection of data of known quality.
Based on EPA's experience in conducting test programs to evaluate the procedures and performance criteria included in Method 30A, EPA recognizes that although prototypes of all equipment needed to perform this method have been successfully demonstrated in the field, at present the equipment needed to follow all procedures required by the method is commercially available only on a limited basis, and is being further refined. One of the issues of greatest concern in the development of an instrumental reference method for Hg has been the design of the sampling probe. Most of the commercially-available probes suitable for Hg measurement are very heavy (over 100 lbs.) making it difficult to move the probe from point-to-point and port-to-port for Hg stratification testing and/or sample traverses. Much progress is being made in probe redesign. One manufacturer has recently developed a probe that weighs less than 40 lbs., samples at significantly lower flow rates, and is suitable for dynamic spiking. Additional field testing of this probe and others currently under development is underway, and EPA plans to continue to actively encourage equipment development and evaluation. To encourage the use of Method 30A, including further development of the supporting equipment, which we believe will eventually enable source testers to perform Hg monitoring system RATAs more efficiently and will become the reference method of choice for many testing companies and affected sources, we are deferring the requirement for implementation of the dynamic spiking and Hg stratification test procedures until January 1, 2009. EPA believes this deferral is reasonable because Hg monitoring data reported to EPA in 2009 will not be used in the trading of Hg allowances, as allowance accounting under the CAMR does not begin until 2010. Source testers are encouraged to use this time to acquire the necessary equipment and familiarize themselves with these procedures. Also, for all emissions test programs and RATAs performed under CAMR prior to January 1, 2009, we are allowing either: (1) A 12-point traverse for sulfur dioxide (SO2) to be substituted for a 12-point Hg traverse, in cases where stratification testing is used to determine the appropriate number and location of the reference method sampling points, or (2) use of the alternate three-point traverse line (0.4, 1.2, and 2.0 meters from the stack wall) as specified in section 8.1.3.2 of Performance Specification 2 (40 CFR part 60, appendix B). We Start Printed Page 51497believe that in the short-term, these temporary deferrals will encourage the application of Method 30A and will help affected CAMR sources meet the January 1, 2009 deadline for initial certification of the required Hg monitoring systems. Several additional Method 30A development considerations are worthy of note. A preliminary draft of Method 30A was first available for public consideration on an EPA Web site (www.epa.gov/ttn/emc/) on February 28, 2006. Since that time, EPA and several stakeholder groups have evaluated the various technical aspects of the method. Based on the combined laboratory and field observations, EPA has been able to simplify several procedural requirements that we believe are essential to the method. The dynamic spiking requirement (for test program-specific verification of measurement system data quality) has been reduced to only a pretest requirement. The interference test has been made optional. The three-point system calibration error test using Hg+2 has been streamlined to a system integrity check using a zero gas and a single upscale Hg+2 gas. Another change has been to relax the Hg0 calibration error specification from 2 percent to 5 percent of span, in recognition of the fact that this procedure is a check of the entire measurement system, as well as the current knowledge regarding the uncertainty of NIST traceable standards. EPA does plan, however, to reconsider this specification relaxation as more field data become available. A final consideration in development of Method 30A has been the requirement for calibration with both Hg0 and Hg+2. Some stakeholders have recommended that we eliminate the Hg0 calibration and rely solely on the Hg+2 calibration. EPA, however, believes this approach would not be adequate, because if only Hg+2 were used, instrument calibration response adjustment could compensate for an unknown amount of converter inefficiency, which would then result in an inaccurate total mercury measurement in situations where Hg0 is an appreciable fraction of the total stack gas Hg.
The second method being added to appendix A-8 of 40 CFR part 60 today is titled “Method 30B—Use of Sorbent Traps to Measure Total Vapor Phase Mercury Emissions from Coal-Fired Combustion Sources.” In Method 30B, a sample of the effluent gas is continuously drawn through a series of tubes containing activated carbon or another sorbent material. After sampling, the tubes are sealed. The Hg captured by the sorbent is then either: (1) Thermally desorbed and analyzed; or (2) the tubes are transferred to a laboratory for extraction of Hg and analysis. Like Method 30A, Method 30B is a performance-based method and contains performance specifications and procedures for hardware selection and calibration, sorbent spiking, and analytical recovery/analysis which allow for development and application of new, improved, and more cost-effective Hg measurement technologies while still ensuring the collection of data of known quality. In particular, Method 30B contains five key measurement performance tests designed to ensure: (1) Selection of a sorbent and analytical technique combination capable of quantitative collection and analysis of gaseous Hg, (2) collection during field testing of enough Hg on each sorbent trap to be reliably quantified, and (3) adequate performance of the method for each test program.
In considering development of a sorbent trap-based reference method, EPA has reviewed historical emissions data where sorbent trap measurement systems were operated concurrently with either the Ontario Hydro Method or Method 29 (40 CFR part 60, appendix A-8). EPA has also conducted several field test evaluations of sorbent trap systems versus the Ontario Hydro Method in collaboration with the Electric Power Research Institute (EPRI). Based on these efforts, we have concluded that a sorbent trap-based technique coupled with appropriate performance criteria and QA procedures can provide Hg emissions data of quality comparable to that produced by the Ontario Hydro Method. Data supporting this conclusion are presented in the docket, EPA-HQ-OAR-2007-0164.
As we have done for Method 30A, for Method 30B emission tests and RATAs performed prior to January 1, 2009, we are allowing either: (1) A 12-point traverse for sulfur dioxide (SO2) to be substituted for a 12-point Hg traverse for the stratification testing used to determine the number and location of the reference method sampling points, or (2) use of the alternate three-point traverse line (0.4, 1.2, and 2.0 meters from the stack wall) as specified in section 8.1.3.2 of Performance Specification 2 (40 CFR part 60, appendix B). We also intend to extend this temporary deferral of mercury stratification testing to application of the Ontario Hydro Method and Method 29. EPA believes this deferral is reasonable because Hg monitoring data reported to EPA in 2009 will not be used in the trading of Hg allowances, as allowance accounting under the CAMR does not begin until 2010.
This direct final rule also amends Performance Specification 12A of appendix B to part 60 by adding Methods 30A and 30B to the list of reference methods acceptable for relative accuracy testing of Hg emissions monitoring systems. Once this direct final rule becomes effective, the reference methods acceptable for Hg measurement in Performance Specification 12A will include Methods 29, 30A, 30B, and ASTM D6784-02.
With today's action, EPA is taking the opportunity to include several considered revisions to the Hg emission monitoring provisions of 40 CFR parts 72 and 75 as described in detail below. EPA is including these revisions in this direct final rule because we believe that they will facilitate implementation of the Hg monitoring under CAMR.
First, § 75.81(a) is being revised to confirm that the Hg CEMS and sorbent trap monitoring systems required under subpart I of part 75 are to measure the total vapor phase mass concentration of Hg in the flue gas, including both the elemental and oxidized forms of Hg, expressed in units of micrograms per standard cubic meter (μg/scm). Although it is generally understood that total vapor phase Hg is the regulated pollutant under CAMR, it recently was brought to EPA's attention that subpart I of part 75 does not explicitly state that Hg monitoring systems only need to measure total vapor phase Hg. The amended language in § 75.81(a) clarifies this.
Second, paragraph (i) in § 75.15 is being revised and a new paragraph (d)(2)(ix) is being added to § 75.20, to codify the rules for using optional non-redundant (“cold”) backup Hg monitoring systems and like-kind replacement Hg analyzers, when the primary Hg monitoring system is unable to provide quality-assured data. For the other types of monitoring systems required by part 75, these monitoring options have been in place since May 1999 (see 64 FR 28597, May 26, 1999). Today's action simply extends these provisions to Hg monitoring systems. Through the years, the regulated community has found these backup monitoring options to be beneficial, in that they minimize the use of missing data substitution procedures during outages of the primary monitoring system.
In particular, § 75.20(d)(2)(ix) specifies that a non-redundant backup Hg monitoring system can either be a Hg CEMS or a sorbent trap monitoring system. The non-redundant backup Hg Start Printed Page 51498monitoring system must be initially certified at each unit or stack location where it will be used, in accordance with § 75.20(d)(2)(i). For a non-redundant backup Hg CEMS, all of the initial certification tests specified in § 75.20(c)(1) are required, except for the 7-day calibration error test. However, for ongoing quality assurance (QA), a RATA is required only once every two years (8 calendar quarters), as specified in § 75.20(d)(2)(vi). For a non-redundant backup sorbent trap monitoring system, a RATA is required for initial certification, and once every two years thereafter for ongoing QA.
When a certified non-redundant backup Hg CEMS or a like-kind replacement Hg analyzer is brought into service, a three-point linearity check with elemental Hg standards and a single-point system integrity check will be required. Alternatively, a three-level system integrity check may be performed instead of these two tests. When a certified non-redundant backup sorbent trap monitoring system is brought into service, only the routine sampling and QA procedures of § 75.15 and appendix K of part 75 will be required.
Each non-redundant backup Hg monitoring system and each like-kind replacement Hg analyzer will be subject to the applicable ongoing QA requirements, restrictions and conditions specified in § 75.20(d)(2). For certified non-redundant backup Hg CEMS and like-kind replacement Hg analyzers, the weekly system integrity checks described in section 2.6 of appendix B of 40 CFR part 75 will also be required as long as the system or analyzer remains in service, unless the daily calibration error tests of the analyzer are done using NIST-traceable oxidized Hg standards.
Third, a new paragraph (k) is being added to § 75.15 that: (1) Clarifies that, when the RATA of an appendix K sorbent trap monitoring system is performed, the type of sorbent material used in the appendix K sorbent traps must be the same as that used for daily operation of the appendix K monitoring system, and (2) allows the appendix K traps used during RATA testing to be smaller than the traps used for daily operation of the appendix K monitoring system. This change will be particularly advantageous at very low Hg concentrations as it will facilitate shorter RATA test run times. Parallel changes are being made to section 6.5.7 of appendix A of part 75 to be consistent with the provisions of § 75.15(k). Section 6.5.7 currently requires the appendix K sorbent traps used for the RATA to be the same size as the traps used for daily operation of the appendix K monitoring system.
Fourth, today's action revises a number of sections of part 75, appendix K, pertaining to the use of sorbent trap monitoring systems. EPA is withdrawing the requirement to use the percentage recovery of the elemental Hg spike in section 3 of each sorbent trap to adjust or “normalize” the Hg mass collected in sections 1 and 2 of the trap. The requirement to spike the third section of each trap is being retained and data from each pair of traps must still be invalidated if either or both spike recovery percentages fall outside the acceptable limits;[1] however, the results of the spike recoveries will no longer be used to adjust the Hg mass collected in the first two sections of the traps. EPA is making this rule change based on an analysis of recent spike recovery data from long-term appendix K field demonstrations. Although the vast majority of the spike recoveries in these studies have been within the currently acceptable limits of 75 to 125 percent, the requirement to normalize based on spike recovery could affect data precision. For a given pair of traps, if one spike recovery was high (e.g., 110 percent) and the other one low (e.g., 90 percent), normalization of the Hg mass collected in the first two trap sections using third section spike recoveries could make it difficult for a pair of sorbent traps to meet the relative deviation (RD) specifications in appendix K. In the example cited, normalization of the data would cause the Hg concentrations measured by the traps to be adjusted by 10 percent in opposite directions, i.e., one upward and one downward. Thus, two Hg concentrations that may have been in close agreement without normalization now might not be able to meet the RD specifications. In view of this, EPA has concluded that evaluating the spike recovery data on a pass/fail basis instead of using the percent recovery values to adjust the emissions data is more technically sound and is also consistent with the way in which the results of daily and quarterly QA assessments of CEMS are interpreted.
Regarding the range of acceptable third section spike recoveries, EPA is not changing the 75 to 125 percent acceptance criteria. As previously noted, early field experience with appendix K monitoring systems has demonstrated that spike recoveries within this range are achievable. However, recent appendix K data indicate that more stringent acceptance criteria may be justifiable. It appears that there has been a marked improvement in third section spike recovery percentages. Recoveries in the range from 85 to 115 percent are consistently being achieved. If this trend continues, EPA may propose to tighten the spike recovery acceptance criteria in a future rulemaking. Toward that end, EPA will continue to collect and evaluate third section spike recovery data from appendix K monitoring systems in the months ahead.
To effect these changes to appendix K, section 11.5 is being removed and reserved; section 10.4 is being revised; Equations K-6 and K-7 are being redesignated as Equations K-5 and K-6, respectively; and the definition of “M*” in redesignated Equation K-5 is being revised.
EPA is also revising appendix K to allow the owner or operator to use other types of gas flow meters besides the conventional dry gas meter (DGM) to quantify sample gas volume. Since the publication of appendix K (see 70 FR 28695, May 18, 2005), numerous requests have been received from the regulated community to allow this flexibility. In response to these requests, EPA initiated an investigation of the feasibility of replacing the DGM in a sorbent trap monitoring system with a thermal mass flow meter. As a result of its investigation, EPA has concluded that a properly calibrated thermal mass flow meter can be at least as accurate as a DGM. The mass flow meter is also a more modern technology than the DGM; since it has no moving parts, it may be more reliable than a DGM for continuous duty.
Having found one type of gas flow meter that can measure as accurately as a DGM, EPA is persuaded that there may be other commercially available gas flow meter technologies that are equally capable and may be suitable for appendix K applications. Accordingly, EPA has decided that a performance-based approach, rather than a prescriptive one, is more appropriate for appendix K gas flow meters. Today's action allows the use of any type of gas flow meter that is capable of accurately measuring gas volumes to within 2 percent.
Section 9.2.2.1 of appendix K now requires the manufacturer of the gas flow meter to perform all necessary set-Start Printed Page 51499up, testing, programming, etc. of the meter and to provide any necessary instructions so that for the particular field application, the meter will give an accurate readout of dry gas volume in units of standard cubic meters. Then, prior to its initial use, the flow meter must be calibrated at a minimum of three settings covering the expected range of sample flow rates for the appendix K system. The initial calibration may be performed either by the manufacturer or by the end user. The calibration of the gas flow meter must be checked quarterly thereafter, at an intermediate flow rate. For mass flow meters, the initial three-point calibration must be performed by using either a compressed gas mixture containing CO2, O2, and N2 in proportions representative of the stack gas composition or by using the actual stack gas. However, when the initial calibration is done with a compressed gas mixture, the mass flow meter may not be used until an additional on-site calibration check of the flow meter at an intermediate flow rate is performed and passed, using the actual stack gas.
To calibrate the gas flow meter, the owner or operator may either follow the basic procedures in section 10.3 or section 16 of Method 5 in appendix A-3 of part 60 for calibration of dry gas meters, or alternatively, may temporarily install a reference gas flow meter (RGFM) at the discharge of the appendix K monitoring system while the monitoring system is in operation and make concurrent measurements of dry stack gas volume with the RGFM and the appendix K gas flow meter. If the latter option is chosen, the RGFM may either be a gas flow metering device that has been calibrated according to section 10.3.1 or section 16 of Method 5 or a NIST-traceable volumetric calibration device with an accuracy of ±1 percent. Note that this alternative calibration technique allows required QA checks to be performed with little or no disruption of the operation of the sorbent trap monitoring system.
Regardless of which calibration approach is used, a calibration factor, Yi, must be obtained at each tested flow rate, where Yi is the ratio of the volume measured by the reference meter to the volume measured by the flow meter being calibrated. For the initial three-point calibration, the three Yi values must be averaged, and each individual Yi must be within ± 0.02 of the average value. The average value, Y, must then be used to correct the gas volumes measured by the gas flow meter. For single-level calibration checks (e.g., the quarterly checks performed for routine QA), the Yi value obtained at the tested flow rate must be compared with the current value of Y. If Yi differs from Y by more than 5 percent, a full three-point recalibration is then required to determine a new Y value.
In this direct final action, the majority of the revised rule provisions pertaining to gas flow meters can be found in sections 5.1.5 and 9.2 of appendix K. Minor revisions to sections 7.2.3 and 7.2.5, Figure K-1, and Table K-1 are being made to be consistent with the changes to sections 5.1.5 and 9.2. In several other places throughout part 75 and in the definition of “Sorbent trap monitoring system” in part 72, the term “dry gas meter,” when used in reference to a sorbent trap monitoring system, is being replaced with the more general term “gas flow meter.” Revisions to section 1.5.2 of appendix B of part 75 will require the gas flow meter calibration procedures and protocols for periodic recalibration of reference gas flow meters to be included in the QA plan for the affected unit.
This direct final action, which approves the use of two optional methods (Methods 30A and 30B) for determining total vapor phase Hg emissions from stationary sources, is being taken in response to numerous public comments concerning the desirability of allowing the use of these types of methods to comply with the Hg emission monitoring requirements of the CAMR for electric utility steam generating units. In the May 18, 2005 final rule (70 FR 28636), we summarized the public comments that we received regarding the use of an instrumental method as an alternative to the Ontario Hydro Method specified in the proposed CAMR. As noted earlier in this preamble, the commenters primarily objected to the required use of the Ontario Hydro Method as the reference method for the RATAs of Hg monitoring systems and expressed concern about the complexities in the method and the amount of time that is required to perform the testing and to receive the results. Commenters pointed out that it could take days to complete the testing and weeks to receive the results from a laboratory. Commenters claimed that for the cap and trade program proposed under CAMR, these delays could lead to significant implementation problems with respect to the timely reporting of emissions data. Further, if a RATA should be failed or invalidated (e.g., if fewer than nine test runs meet the relative deviation criterion for the paired Ontario Hydro trains), data from the Hg monitoring system would be invalidated from the hour of the failed or invalidated test until the hour of completion of a successful RATA. Conservatively high substitute data values would have to be reported during that entire time period. In our response to those comments in the final CAMR rule, we stated that the alternative use of an instrumental method for the required RATAs of Hg monitoring systems and sorbent trap monitoring systems is allowed by the final rule but is subject to approval by the Administrator. We also stated our commitment to propose and promulgate a Hg instrumental reference method once sufficient supporting field test data become available. We further stated that “A Hg instrumental reference method for RATA testing is vastly preferable to the Ontario Hydro Method and will greatly facilitate the implementation of a Hg cap-and-trade program.”
Since promulgation of CAMR, we have continued to communicate with stakeholders interested in the Hg monitoring requirements of the rule, and we have come to more clearly understand that it is of great interest to the affected entities to have additional reference method options available for relative accuracy testing of installed Hg monitoring systems as soon as possible. Accordingly, at the end of 2005, we began developing an instrumental test method for Hg and solicited feedback from the stakeholders on a working draft of the method (referred to as PRE-009 at http://www.epa.gov/ttn/emc/prelim.html). More recently, we have been developing a viable sorbent trap reference method. These efforts have resulted in Methods 30A and 30B.
The general beneficial impacts of this direct final rule to approve the two optional Hg test methods and amend targeted portions of 40 CFR parts 72 and 75 include: Allowing affected sources to choose the use of an alternative to the Ontario Hydro Method without the administrative burden of applying for Administrator approval on a case-by-case basis; providing the availability of real-time RATA results (Method 30A); reducing the overall RATA testing times; reducing costs relative to the Ontario Hydro Method; and providing additional flexibility in appendix K sorbent trap monitoring and backup monitoring approaches. The two optional methods being approved by this direct final rule are considered to be comparable to the Ontario Hydro Method in terms of the quality of the results produced. Over the last year, EPA has collaborated with EPRI and some of its members in a number of field test programs that have confirmed that the instrumental reference method approved/established in this notice will provide data comparable to or better Start Printed Page 51500than that of the “Ontario Hydro Method.”
Assuming we do not receive adverse comment on this direct final rulemaking and Methods 30A and 30B become final, we plan to post information relevant to Method 30A and 30B applications and equipment advances on EPA's Web site at http://www.epa.gov/airmarkets.
VII. Statutory and Executive Order Reviews
A. Executive Order 12866: Regulatory Planning and Review
This action is not a “significant regulatory action” under the terms of Executive Order (EO) 12866 (58 FR 51735, October 4, 1993) and is therefore not subject to review under the EO.
B. Paperwork Reduction Act
This action does not impose an information collection burden under the provisions of the Paperwork Reduction Act, 44 U.S.C. 3501 et seq. Burden means the total time, effort, or financial resources expended by persons to generate, maintain, retain, or disclose or provide information to or for a Federal agency. This includes the time needed to review instructions; develop, acquire, install, and utilize technology and systems for the purposes of collecting, validating, and verifying information, processing and maintaining information, and disclosing and providing information; adjust the existing ways to comply with any previously applicable instructions and requirements; train personnel to be able to respond to a collection of information; search data sources; complete and review the collection of information; and transmit or otherwise disclose the information.
An agency may not conduct or sponsor, and a person is not required to respond to a collection of information, unless it displays a currently valid OMB control number. The OMB control numbers for EPA's regulations in 40 CFR are listed in 40 CFR part 9.
C. Regulatory Flexibility Act
The Regulatory Flexibility Act (RFA) generally requires an agency to prepare a regulatory flexibility analysis of any rule subject to notice and comment rulemaking requirements under the Administrative Procedure Act or any other statute unless the agency certifies that the rule will not have a significant economic impact on a substantial number of small entities. Small entities include small businesses, small organizations, and small governmental jurisdictions.
For purposes of assessing the impacts of today's rule on small entities, small entity is defined as: (1) A small business whose parent company has fewer than 100 or 1,000 employees, or fewer than 4 billion kilowatt-hr per year of electricity usage, depending on the size definition for the affected North American Industry Classification System code; (2) a small governmental jurisdiction that is a government of a city, county, town, school district or special district with a population of less than 50,000; and (3) a small organization that is any not-for-profit enterprise which is independently owned and operated and is not dominant in its field.
After considering the economic impacts of today's direct final rule on small entities, I certify that this action will not have a significant economic impact on a substantial number of small entities. This direct final rule will not impose any requirements on small entities because it does not impose any additional regulatory requirements, but rather provides clarification and additional regulatory flexibilty.
D. Unfunded Mandates Reform Act
Title II of the Unfunded Mandates Reform Act of 1995 (UMRA), Pub. L. 104-4, establishes requirements for Federal agencies to assess the effects of their regulatory actions on State, local, and tribal governments and the private sector. Under section 202 of the UMRA, EPA generally must prepare a written statement, including a cost-benefit analysis, for proposed and final rules with “Federal mandates” that may result in expenditures to State, local, and tribal governments, in the aggregate, or to the private sector, of $100 million or more in any one year. Before promulgating an EPA rule for which a written statement is needed, section 205 of the UMRA generally requires EPA to identify and consider a reasonable number of regulatory alternatives and adopt the least costly, most cost-effective or least burdensome alternative that achieves the objectives of the rule. The provisions of section 205 do not apply when they are inconsistent with applicable law. Moreover, section 205 allows EPA to adopt an alternative other than the least costly, most cost-effective, or least burdensome alternative if the Administrator publishes with the final rule an explanation why that alternative was not adopted. Before EPA establishes any regulatory requirements that may significantly or uniquely affect small governments, including tribal governments, it must have developed under section 203 of the UMRA a small government agency plan. The plan must provide for notifying potentially affected small governments, enabling officials of affected small governments to have meaningful and timely input in the development of EPA regulatory proposals with significant Federal intergovernmental mandates, and informing, educating, and advising small governments on compliance with the regulatory requirements.
EPA has determined that this direct final rule does not contain a Federal mandate that may result in expenditures of $100 million or more for State, local, and tribal governments in the aggregate, or to the private sector in any 1 year, nor does this rule significantly or uniquely impact small governments, because it contains no requirements that impose new obligations upon them. Thus, this direct final rule is not subject to the requirements of sections 202 and 205 of the UMRA.
E. Executive Order 13132: Federalism
Executive Order 13132, entitled “Federalism” (64 FR 43255, August 10, 1999), requires EPA to develop an accountable process to ensure “meaningful and timely input by State and local officials in the development of regulatory policies that have federalism implications.” “Policies that have federalism implications” is defined in the Executive Order to include regulations that have “substantial direct effects on the States, on the relationship between the national government and the States, or on the distribution of power and responsibilities among the various levels of government.”
This direct final rule does not have federalism implications. It will not have substantial direct effects on the States, on the relationship between the national government and the States, or on the distribution of power and responsibilities among the various levels of government, as specified in Executive Order 13132. The use of these methods is optional on the part of the regulated entities listed. Thus, Executive Order 13132 does not apply to this direct final rule.
F. Executive Order 13175: Consultation and Coordination With Indian Tribal Governments
Executive Order 13175, entitled “Consultation and Coordination with Indian Tribal Governments” (65 FR 67249, November 9, 2000), requires EPA to develop an accountable process to ensure “meaningful and timely input by tribal officials in the development of regulatory policies that have tribal implications.” This direct final rule does not have tribal implications, as specified in Executive Order 13175. It will not have substantial direct effects on tribal governments, on the Start Printed Page 51501relationship between the Federal government and Indian tribes, or on the distribution of power and responsibilities between the Federal government and Indian tribes. Thus, Executive Order 13175 does not apply to this final rule.
G. Executive Order 13045: Protection of Children From Environmental Health and Safety Risks
Executive Order 13045: “Protection of Children from Environmental Health Risks and Safety Risks” (62 FR 19885, April 23, 1997) applies to any rule that: (1) Is determined to be “economically significant” as defined under Executive Order 12866, and (2) concerns an environmental health or safety risk that EPA has reason to believe may have a disproportionate effect on children. If the regulatory action meets both criteria, the Agency must evaluate the environmental health or safety effects of the planned rule on children, and explain why the planned regulation is preferable to other potentially effective and reasonably feasible alternatives considered by the Agency. EPA interprets Executive Order 13045 as applying only to those regulatory actions that are based on health or safety risks, such that the analysis required under section 5-501 of the Order has the potential to influence the regulation. This rule is not subject to Executive Order 13045 because it does not establish an environmental standard intended to mitigate health or safety risks.
H. Executive Order 13211: Actions That Significantly Affect Energy Supply, Distribution, or Use
This rule is not subject to Executive Order 13211, “Actions Concerning Regulations That Significantly Affect Energy Supply, Distribution, or Use” (66 FR 28355, May 22, 2001) because it is not a significant regulatory action under Executive Order 12866.
I. National Technology Transfer Advancement Act
Section 12(d) of the National Technology Transfer and Advancement Act of 1995 (NTTAA), Public Law No. 104-113, section 12(d) (15 U.S.C. 272 note) directs EPA to use voluntary consensus standards in its regulatory activities unless to do so would be inconsistent with applicable law or otherwise impractical. Voluntary consensus standards are technical standards (e.g., materials specifications, test methods, sampling procedures, and business practices) that are developed or adopted by voluntary consensus standards bodies. The NTTAA directs EPA to provide Congress, through OMB, explanations when the Agency decides not to use available and applicable voluntary consensus standards. This rulemaking involves technical standards. Consistent with the NTTAA, EPA in a previous related rulemaking (70 FR 28606, May 18, 2005) identified an acceptable VCS for measuring Hg emissions. The standard ASTM D6784-02, Standard Test Method for Elemental, Oxidized, Particle-Bound and Total Mercury Gas Generated from Coal-Fired Stationary sources (Ontario Hydro Method) was cited in that final rule for measuring Hg emissions. After today's action becomes effective, the Ontario Hydro Method will remain an acceptable method for measuring Hg emissions.
J. Executive Order 12898: Federal Actions To Address Environmental Justice in Minority Populations and Low-Income Populations
Executive Order 12898 (59 FR 7629 (Feb. 16, 1994)) establishes federal executive policy on environmental justice. Its main provision directs federal agencies, to the greatest extent practicable and permitted by law, to make environmental justice part of their mission by identifying and addressing, as appropriate, disproportionately high and adverse human health or environmental effects of their programs, policies, and activities on minority populations and low-income populations in the United States.
EPA has determined that this direct final rule will not have disproportionately high and adverse human health or environmental effects on minority or low-income populations because it does not affect the level of protection provided to human health or the environment. This direct final rule does not affect or relax the control measures on sources impacted by this rule and therefore will not cause emissions increases from these sources.
K. Congressional Review Act
The Congressional Review Act, 5 U.S.C. 801 et seq., as added by the Small Business Regulatory Enforcement Fairness Act of 1996, generally provides that before a rule may take effect, the Agency promulgating the rule must submit a rule report, which includes a copy of the rule, to each House of the Congress and to the Comptroller General of the United States. EPA will submit a report containing this rule and other required information to the U.S. Senate, the U.S. House of Representatives, and the Comptroller General of the United States prior to publication of the rule in the Federal Register. A major rule cannot take effect until 60 days after it is published in the Federal Register. This action is not a “major rule” as defined by 5 U.S.C. 804(2). This rule will be effective on November 6, 2007.
Start List of SubjectsList of Subjects
40 CFR Part 60
- Environmental protection
- Administrative practice and procedures
- Air pollution control
- Continuous emission monitors
- Electric utilities
- Mercury
- Test methods and procedures
40 CFR Part 72
- Environmental protection
- Administrative practice and procedures
- Air pollution control
- Continuous emission monitors
- Electric utilities
- Mercury
- Test methods and procedures
40 CFR Part 75
- Environmental protection
- Administrative practice and procedures
- Air pollution control
- Continuous emission monitors
- Electric utilities
- Mercury
- Test methods and procedures
End Signature Start Amendment PartDated: August 17, 2007.
Stephen L. Johnson,
Administrator.
For the reasons set out in the preamble, title 40, chapter I, parts 60, 72, and 75 of the Code of Federal Regulations are amended as follows:
End Amendment Part Start PartPART 60—STANDARDS OF PERFORMANCE FOR NEW STATIONARY SOURCES
End Part Start Amendment Part1. The authority citation for part 60 continues to read as follows:
End Amendment PartAppendix A-8 [Amended]
Start Amendment Part2. Amend Appendix A-8 by revising the heading and adding in numerical order Methods 30A and 30B to read as follows:
End Amendment PartAPPENDIX A-8 TO PART 60—TEST METHODS 26 THROUGH 30B
* * * * *Method 30A—Determination of Total Vapor Phase Mercury Emissions From Stationary Sources (Instrumental Analyzer Procedure)
1.0 Scope and Application
What Is Method 30A?
Method 30A is a procedure for measuring total vapor phase mercury (Hg) emissions from stationary sources using an instrumental analyzer. This method is particularly appropriate for performing emissions testing and for conducting relative accuracy test audits (RATAs) of mercury continuous emissions monitoring systems (Hg CEMS) and sorbent trap monitoring systems at coal-fired combustion sources. Quality assurance and quality control Start Printed Page 51502requirements are included to assure that you, the tester, collect data of known and acceptable quality for each testing site. This method does not completely describe all equipment, supplies, and sampling procedures and analytical procedures you will need but refers to other test methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional methods which are also found in appendices A-1 and A-3 to this part:
(a) Method 1—Sample and Velocity Traverses for Stationary Sources.
(b) Method 4—Determination of Moisture Content in Stack Gases.
1.1 Analytes. What does this method determine? This method is designed to measure the mass concentration of total vapor phase Hg in flue gas, which represents the sum of elemental Hg (Hg0) and oxidized forms of Hg (Hg+2), in mass concentration units of micrograms per cubic meter (μg/m3).
Analyte CAS No. Sensitivity Elemental Hg (Hg0) 7439-97-6 Typically <2% of Calibration Span. Oxidized Hg (Hg+2) (Same). 1.2 Applicability. When is this method required? Method 30A is offered as a reference method for emission testing and for RATAs of Hg CEMS and sorbent trap monitoring systems at coal-fired boilers. Method 30A may also be specified for other source categories in the future, either by New Source Performance Standards (NSPS), National Emission Standards for Hazardous Air Pollutants (NESHAP), emissions trading programs, State Implementation Plans (SIP), or operating permits that require measurement of Hg concentrations in stationary source emissions to determine compliance with an applicable emission standard or limit, or to conduct RATAs of Hg CEMS and sorbent trap monitoring systems.
1.3 Data Quality Objectives (DQO). How good must my collected data be? Method 30A has been designed to provide data of high and known quality for Hg emission testing and for relative accuracy testing of Hg monitoring systems including Hg CEMS and sorbent trap monitoring systems. In these and other applications, the principle objective is to ensure the accuracy of the data at the actual emission levels encountered. To meet this objective, calibration standards prepared according to an EPA traceability protocol must be used and measurement system performance tests are required.
2.0 Summary of Method
In this method, a sample of the effluent gas is continuously extracted and conveyed to an analyzer capable of measuring the total vapor phase Hg concentration. Elemental and oxidized mercury (i.e., Hg0 and Hg+2) may be measured separately or simultaneously but, for purposes of this method, total vapor phase Hg is the sum of Hg0 and Hg+2. You must meet the performance requirements of this method (i.e., system calibration, interference testing, dynamic spiking, and system integrity/drift checks) to validate your data. The dynamic spiking requirement is deferred until January 1, 2009.
3.0 Definitions
3.1 Calibration Curve means the relationship between an analyzer's response to the injection of a series of calibration gases and the actual concentrations of those gases.
3.2 Calibration Gas means a gas standard containing Hg0 or HgCl2 at a known concentration that is produced and certified in accordance with an EPA traceability protocol for certification of Hg calibration standards.
3.2.1 Zero Gas means a calibration gas with a concentration that is below the level detectable by the measurement system.
3.2.2 Low-Level Gas means a calibration gas with a concentration that is 10 to 30 percent of the calibration span.
3.2.3 Mid-Level Gas means a calibration gas with a concentration that is 40 to 60 percent of the calibration span.
3.2.4 High-Level Gas means a calibration gas whose concentration is equal to the calibration span.
3.3 Converter means a device that reduces oxidized mercury (Hg+2) to elemental mercury (Hg0).
3.4 Calibration Span means the upper limit of valid instrument response during sampling. To the extent practicable the measured emissions are to be between 10 and 100 percent of the selected calibration span (i.e., the measured emissions should be within the calibrated range determined by the Low- and High-Level gas standards). It is recommended that the calibration span be at least twice the native concentration to accommodate the dynamic spiking procedure.
3.5 Centroidal Area means the central area that has the same shape as the stack or duct cross section and is no greater than one percent of the stack or duct total cross-sectional area.
3.6 Data Recorder means the equipment that permanently records the concentrations reported by the analyzer.
3.7 Drift Check means the test to determine the difference between the measurement system readings obtained in a post-run system integrity check and the prior pre-run system integrity check at a specific calibration gas concentration level (i.e., zero, mid-level, or high-level).
3.8 Dynamic Spiking means a procedure in which a known mass or concentration of vapor phase HgCl2 is injected into the probe sample gas stream at a known flow rate, in order to assess the effects of the flue gas matrix on the accuracy of the measurement system.
3.9 Gas Analyzer means the equipment that detects the total vapor phase Hg being measured and generates an output proportional to its concentration.
3.10 Interference Test means the test to detect analyzer responses to compounds other than Hg, usually gases present in the measured gas stream, that are not adequately accounted for in the calibration procedure and may cause measurement bias.
3.11 Measurement System means all of the equipment used to determine the Hg concentration. The measurement system may generally include the following major subsystems: sample acquisition, Hg+2 to Hg0 converter, sample transport, sample conditioning, flow control/gas manifold, gas analyzer, and data recorder.
3.12 Native Concentration means the total vapor phase Hg concentration in the effluent gas stream.
3.13 NIST means the National Institute of Standards and Technology, located in Gaithersburg, Maryland.
3.14 Response Time means the time it takes for the measurement system, while operating normally at its target sample flow rate or dilution ratio, to respond to a known step change in gas concentration (from a low-level to a high-level gas) and to read within 5 percent of the stable high-level gas response.
3.15 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.16 System Calibration Error means the difference between the measured concentration of a low-, mid-, or high-level Hg0 calibration gas and the certified concentration of the gas when it is introduced in system calibration mode.
3.17 System Calibration Mode means introducing the calibration gases into the measurement system at the probe, upstream of all sample conditioning components.
3.18 Test refers to the series of runs required by the applicable regulation.
4.0 Interferences
Interferences will vary among instruments and potential instrument-specific spectral and matrix interferences must be evaluated through the interference test and the dynamic spiking tests.
5.0 Safety
What safety measures should I consider when using this method?
This method may require you to work with hazardous materials and in hazardous conditions. You are encouraged to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user's manual. Occupational Safety and Health Administration (OSHA) regulations concerning use of compressed gas cylinders and noxious gases may apply. Start Printed Page 51503
6.0 Equipment and Supplies
6.1 What do I need for the measurement system? This method is intended to be applicable to multiple instrumental technologies. You may use any equipment and supplies that meet the following specifications.
6.1.1 All wetted sampling system components, including probe components prior to the point at which the calibration gas is introduced, must be chemically inert to all Hg species. Materials such as perfluoroalkoxy (PFA) TeflonTM, quartz, treated stainless steel (SS) are examples of such materials. [Note: These materials of construction are required because components prior to the calibration gas injection point are not included in the system calibration error, system integrity, and interference tests.]
6.1.2 The interference, system calibration error, system integrity, drift and dynamic spiking test criteria must all be met by the system used.
6.1.3 The system must be capable of measuring and controlling sample flow rate.
6.1.4 All system components prior to the Hg+2 to Hg0 converter must be maintained at a sample temperature above the acid gas dew point.
6.2 Measurement System Components. Figure 30A-1 in Section 17.0 is an example schematic of a Method 30A measurement system.
6.2.1 Sample Probe. The probe must be made of the appropriate materials as noted in Section 6.1.1, heated when necessary (see Section 6.1.4), configured with ports for introduction of calibration and spiking gases, and of sufficient length to traverse all of the sample points.
6.2.2 Filter or Other Particulate Removal Device. The filter or other particulate removal device is considered to be a part of the measurement system, must be made of appropriate materials as noted in Section 6.1.1, and must be included in all system tests.
6.2.3 Sample Line. The sample line that connects the probe to the converter, conditioning system and analyzer must be made of appropriate materials as noted in Section 6.1.1.
6.2.4 Conditioning Equipment. For dry basis measurements, a condenser, dryer or other suitable device is required to remove moisture continuously from the sample gas. Any equipment needed to heat the probe, or sample line to avoid condensation prior to the moisture removal component is also required. For wet basis systems, you must keep the sample above its dew point either by: (1) Heating the sample line and all sample transport components up to the inlet of the analyzer (and, for hot-wet extractive systems, also heating the analyzer) or (2) by diluting the sample prior to analysis using a dilution probe system. The components required to do either of the above are considered to be conditioning equipment.
6.2.5 Sampling Pump. A pump is needed to push or pull the sample gas through the system at a flow rate sufficient to minimize the response time of the measurement system. If a mechanical sample pump is used and its surfaces are in contact with the sample gas prior to detection, the pump must be leak free and must be constructed of a material that is non-reactive to the gas being sampled (see Section 6.1.1). For dilution-type measurement systems, an ejector pump (eductor) may be used to create a sufficient vacuum that sample gas will be drawn through a critical orifice at a constant rate. The ejector pump may be constructed of any material that is non-reactive to the gas being sampled.
6.2.6 Calibration Gas System(s). One or more systems may be needed to introduce calibration gases into the measurement system. A system should be able to flood the sampling probe sufficiently to prevent entry of gas from the effluent stream.
6.2.7 Dynamic Spiking Port. For the purposes of the dynamic spiking procedure described in Section 8.2.7, the measurement system must be equipped with a port to allow introduction of the dynamic spike gas stream with the sample gas stream, at a point as close as possible to the inlet of the probe so as to ensure adequate mixing. The same port used for system calibrations and calibration error checks may be used for dynamic spiking purposes.
6.2.8 Sample Gas Delivery. The sample line may feed directly to a converter, to a by-pass valve (for speciating systems), or to a sample manifold. All valve and/or manifold components must be made of material that is non-reactive to the gas sampled and the calibration gas, and must be configured to safely discharge any excess gas.
6.2.9 Hg Analyzer. An instrument is required that continuously measures the total vapor phase Hg in the gas stream and meets the applicable specifications in Section 13.0.
6.2.10 Data Recorder. A recorder, such as a computerized data acquisition and handling system (DAHS), digital recorder, strip chart, or data logger, is required for recording measurement data.
6.3 Moisture Measurement System. If correction of the measured Hg emissions for moisture is required (see Section 8.5), either Method 4 in appendix A-3 to this part or other moisture measurement methods approved by the Administrator will be needed to measure stack gas moisture content.
7.0 Reagents and Standards
7.1 Calibration Gases. What calibration gases do I need? You will need calibration gases of known concentrations of Hg0 and HgCl2. Special reagents and equipment may be required to prepare the HgCl2 gas standards (e.g., a NIST-traceable solution of HgCl2 and a gas generator equipped with mass flow controllers).
The following calibration gas concentrations are required:
7.1.1 High-Level Gas. Equal to the selected calibration span.
7.1.2 Mid-Level Gas. 40 to 60 percent of the calibration span.
7.1.3 Low-Level Gas. 10 to 30 percent of the calibration span.
7.1.4 Zero Gas. No detectable Hg.
7.1.5 Dynamic Spike Gas. The exact concentration of the HgCl2 calibration gas used to perform the pre-test dynamic spiking procedure described in Section 8.2.7 depends on the native Hg concentration in the stack The spike gas must produce a spiked sample concentration above the native concentration, as specified in Section 8.2.7.2.2.
7.2 Interference Test. What reagents do I need for the interference test? Use the appropriate test gases listed in Table 30A-3 in Section 17.0 (i.e., the potential interferents for the source to be tested, as identified by the instrument manufacturer) to conduct the interference check. These gases need not be of protocol gas quality.
8.0 Sample Collection
Emission Test Procedure
Figure 30A-2 in Section 17.0 presents an overview of the test procedures required by this method. Since you may choose different options to comply with certain performance criteria, you must identify the specific options and associated frequencies you select and document your results in regard to the performance criteria.
8.1 Sample Point Selection. What sampling site and sampling points do I select?
8.1.1 When this method is used solely for Hg emission testing (e.g., to determine compliance with an emission standard or limit), use twelve sampling points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, you may conduct a stratification test as described in Section 8.1.3 to determine the number and location of the sampling points.
8.1.2 When this method is used for relative accuracy testing of a Hg CEMS or sorbent trap monitoring system, follow the sampling site selection and sampling point layout procedures for gas monitor RATA testing described in the appropriate performance specification or applicable regulation (e.g., Performance Specification 2, section 8.1.3 of appendix B to this part or section 6.5.6 of appendix A to part 75 of this chapter), with one exception. If you elect to perform stratification testing as part of the sampling point selection process, perform the testing in accordance with Section 8.1.3 of this method (see also “Summary Table of QA/QC Requirements” in Section 9.0).
8.1.3 Determination of Stratification. If you elect to perform stratification testing as part of the sampling point selection process and the test results show your effluent gas stream to be unstratified or minimally stratified, you may be allowed to sample at fewer points or at different points than would otherwise be required.
8.1.3.1 Test Procedure. To test for stratification, use a probe of appropriate length to measure the total vapor phase Hg concentration at twelve traverse points located according to Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part. Alternatively, for a sampling location where stratification is expected (e.g., after a wet scrubber or at a point where dissimilar gas streams are combined together), if a 12-point Hg stratification test has been previously performed at that location and the results of the test showed the location to be minimally stratified or unstratified according to the criteria in section 8.1.3.2, you may perform an abbreviated 3-point or 6-point Hg stratification test at the points specified in Start Printed Page 51504section 6.5.6.2(a) of appendix A to part 75 of this chapter in lieu of performing the 12-point test. Sample for a minimum of twice the system response time (see Section 8.2.6) at each traverse point. Calculate the individual point and mean Hg concentrations.
8.1.3.2 Acceptance Criteria and Sampling Point Location.
8.1.3.2.1 If the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a) ±5 percent of the mean concentration; or (b) ±0.2 μg/m3 (whichever is less restrictive), the gas stream is considered to be unstratified and you may collect samples from a single point that most closely matches the mean.
8.1.3.2.2 If the 5 percent or 0.2 μg/m3 criterion in Section 8.1.3.2.1 is not met, but the Hg concentration at each traverse point differs from the mean concentration for all traverse points by no more than: (a)±10 percent of the mean; or (b)±0.5 μg/m3 (whichever is less restrictive), the gas stream is considered to be minimally stratified, and you may take samples from three points, provided the points are located on the measurement line exhibiting the highest average Hg concentration during the stratification test. If the stack diameter (or equivalent diameter, for a rectangular stack or duct) is greater than 2.4 meters (7.8 ft), locate the three sampling points at 0.4, 1.0, and 2.0 meters from the stack or duct wall. Alternatively, if a RATA required by part 75 of this chapter is being conducted, you may locate the three points at 4.4, 14.6, and 29.6 percent of the duct diameter, in accordance with Method 1 in appendix A-1 to this part. For stack or duct diameters of 2.4 meters (7.8 ft) or less, locate the three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line.
8.1.3.2.3 If the gas stream is found to be stratified because the 10 percent or 0.5 μg/m3 criterion in Section 8.1.3.2.2 is not met, then either locate three sampling points at 16.7, 50.0, and 83.3 percent of the measurement line that exhibited the highest average Hg concentration during the stratification test, or locate twelve traverse points for the test in accordance with Table 1-1 or Table 1-2 of Method 1 in appendix A-1 to this part; or, if a RATA required by part 75 of this chapter is being conducted, locate six Method 1 points along the measurement line that exhibited the highest average Hg concentration.
8.1.3.3 Temporal Variations. Temporal variations in the source Hg concentration during a stratification test may complicate the determination of stratification. If temporal variations are a concern, you may use the following procedure to normalize the stratification test data. A second Hg measurement system, i.e., either an installed Hg CEMS or another Method 30A system, is required to perform this procedure. Position the sampling probe of the second Hg measurement system at a fixed point in the stack or duct, at least one meter from the stack or duct wall. Then, each time that the Hg concentration is measured at one of the stratification test points, make a concurrent measurement of Hg concentration at the fixed point. Normalize the Hg concentration measured at each traverse point, by multiplying it by the ratio of CF,avg to CF, where CF is the corresponding fixed-point Hg concentration measurement, and CF,avg is the average of all of the fixed-point measurements over the duration of the stratification test. Evaluate the results of the stratification test according to section 8.1.3.2, using the normalized Hg concentrations.
8.1.3.4 Stratification Testing Exemption. Stratification testing need not be performed at a test location where it would otherwise be required to justify using fewer sample points or different sample points, if the owner or operator documents that the Hg concentration in the stack gas is expected to be 3 μg/m3 or less at the time of a Hg monitoring system RATA or an Hg emissions test. To demonstrate that a particular test location qualifies for the stratification testing exemption, representative Hg emissions data must be collected just prior to the RATA or emissions test. At least one hour of Hg concentration data is required for the demonstration. The data used for the demonstration shall be recorded at process operating conditions that closely approximate the operating conditions that will exist during the RATA or emissions test. It is recommended that collection of the demonstration data be integrated with the on-site pretest procedures required by the reference method being used for the RATA or emissions test (whether this method or another approved Hg reference method is used). Quality-assured data from an installed Hg monitoring system may also be used for the demonstration. If a particular test location qualifies for the stratification testing exemption, sampling shall be performed at three points, as described in section 8.1.3.2.2 of this method. The owner or operator shall fully document the method used to collect the demonstration data and shall keep this documentation on file with the data from the associated RATA or Hg emissions test.
8.1.3.5 Interim Alternative Stratification Test Procedures. In the time period between the effective date of this method and January 1, 2009, you may follow one of the following two procedures. Substitute a stratification test for sulfur dioxide (SO2) for the Hg stratification test described in section 8.1.3.1. If this option is chosen, follow the test procedures in section 6.5.6.1 of appendix A to part 75 of this chapter. Evaluate the test results and determine the sampling point locations according to section 6.5.6.3 of appendix A to part 75 of this chapter. If the sampling location is found to be minimally stratified or unstratified for SO2, it shall be considered minimally stratified or unstratified for Hg. Alternatively, you may forgo stratification testing, assume the gas stream is minimally stratified, and sample at three points as described in section 8.1.3.2.2 of this method.
8.2 Initial Measurement System Performance Tests. What initial performance criteria must my system meet before I begin sampling? Before measuring emissions, perform the following procedures:
(a) Interference Test;
(b) Calibration Gas Verification;
(c) Measurement System Preparation;
(d) 3-Point System Calibration Error Test;
(e) System Integrity Check;
(f) Measurement System Response Time Test; and
(g) Dynamic Spiking Test.
8.2.1 Interference Test (Optional). Your measurement system should be free of known interferences. It is recommended that you conduct this interference test of your measurement system prior to its initial use in the field to verify that the candidate test instrument is free from inherent biases or interferences resulting from common combustion emission constituents. If you have multiple measurement systems with components of the same make and model numbers, you need only perform this interference check on one system and you may also rely on an interference test conducted by the manufacturer on a system having components of the same make and model(s) of the system that you use. The interference test procedure is found in Section 8.6 of this method.
8.2.2 Calibration Gas Verification. How must I verify the concentrations of my calibration gases?
8.2.2.1 Cylinder Gas Standards. When cylinder gas standards are used for Hg0, obtain a certificate from the gas manufacturer and confirm that the documentation includes all information required by an EPA traceability protocol (see Section 16). Confirm that the manufacturer certification is complete and current. Ensure that the calibration gas certifications have not expired.
8.2.2.2 Other Calibration Standards. All other calibration standards for HgCl2 and Hg0, such as gas generators, must meet the requirements of an EPA traceability protocol (see Section 16), and the certification procedures must be fully documented in the test report.
8.2.2.3 Calibration Span. Select the calibration span (i.e., high-level gas concentration) so that the measured source emissions are 10 to 100 percent of the calibration span. This requirement is waived for applications in which the Hg concentrations are consistently below 1 μg/m3; however, the calibration span for these low-concentration applications shall not exceed 5 μg/m3.
8.2.3 Measurement System Preparation. How do I prepare my measurement system for use? Assemble, prepare, and precondition the measurement system according to your standard operating procedure. Adjust the system to achieve the correct sampling rate or dilution ratio (as applicable). Then, conduct a 3-point system calibration error test using Hg0 as described in Section 8.2.4, an initial system integrity check using HgCl2 and a zero gas as described in Section 8.2.5, and a pre-test dynamic spiking test as described in Section 8.2.7.
8.2.4 System Calibration Error Test. Conduct a 3-point system calibration error test before the first test run. Use Hg0 standards for this test. Introduce the low-, mid-, and high-level calibration gases in any order, in system calibration mode, unless you desire to determine the system response time during this test, in which case, inject the gases such that the high-level injection Start Printed Page 51505directly follows the low-level injection. For non-dilution systems, you may adjust the system to maintain the correct flow rate at the analyzer during the test, but you may not make adjustments for any other purpose. For dilution systems, you must operate the measurement system at the appropriate dilution ratio during all system calibration error checks, and you may make only the adjustments necessary to maintain the proper ratio. After each gas injection, wait until a stable response has been obtained. Record the analyzer's final, stable response to each calibration gas on a form similar to Table 30A-1 in Section 17.0. For each calibration gas, calculate the system calibration error using Equation 30A-1 in Section 12.2. The calibration error specification in Section 13.1 must be met for the low-, mid-, and high-level gases. If the calibration error specification is not met for all three gases, take corrective action and repeat the test until an acceptable 3-point calibration is achieved.
8.2.5 System Integrity Check. Perform a two-point system integrity check before the first test run. Use the zero gas and either the mid- or high-level HgCl2 calibration gas for the check, whichever one best represents the total vapor phase Hg concentration levels in the stack. Record the data on a form similar to Table 30A-2 in Section 17.0. The system integrity check specification in Section 13.2 must be met for both the zero gas and the mid- or high-level gas. If the system integrity specification is not met for both gases, take corrective action and repeat the test until an acceptable system integrity check is achieved.
8.2.6 Measurement System Response Time. The measurement system response time is used to determine the minimum sampling time for each sampling point and is equal to the time that is required for the measured Hg concentration to increase from the stable low-level calibration gas response to a value within 5 percent of the stable high-level calibration gas response during the system calibration error test in Section 8.2.4. Round off the measured system response time to the nearest minute.
8.2.7 Dynamic Spiking Test. You must perform dynamic spiking prior to the first test run to validate your test data. The purpose of this procedure is to demonstrate that the site-specific flue gas matrix does not adversely affect the accuracy of the measurement system. The specifications in Section 13.5 must be met to validate your data. If these specifications are not met for the pre-test dynamic spiking, you may not proceed with the test until satisfactory results are obtained. For the time period between the effective date of this method and January 1, 2009, the dynamic spiking requirement is waived.
8.2.7.1 How do I perform dynamic spiking? Dynamic spiking is a gas phase application of the method of standard additions, which involves injecting a known quantity of Hg into the measurement system upstream of all sample conditioning components, similar to system calibration mode, except the probe is not flooded and the resulting sample stream includes both effluent gas and the spike gas. You must follow a written procedure that details how the spike is added to the system, how the spike dilution factor (DF) is measured, and how the Hg concentration data are collected and processed.
8.2.7.2 Spiking Procedure Requirements.
8.2.7.2.1 Spiking Gas Requirements. The spike gas must also be a HgCl2 calibration gas certified by an EPA traceability protocol. You must choose concentrations that can produce the target levels while being injected at a volumetric flow rate that is ≤20 percent of the total volumetric flow rate through the measurement system (i.e., sample flow rate plus spike gas flow rate).
8.2.7.2.2 Target Spiking Level. The target level for spiking must be 150 to 200 percent of the native Hg concentration; however, if the native Hg concentration is <1 μg/m3, set the target level to add between 1 and 4 μg/m3 Hg+2 to the native concentration. Use Equation 30A-5 in Section 12.5 to calculate the acceptable range of spike gas concentrations at the target level. Then select a spike gas concentration in that range.
8.2.7.2.3 Spike Injections. You must inject spikes in such a manner that the spiking does not alter the total volumetric sample system flow rate and dilution ratio (if applicable). You must collect at least 3 data points, and the relative standard deviation (RSD) specification in Section 13.5 must be met. Each data point represents a single spike injection, and pre- and post-injection measurements of the native Hg concentration (or diluted native concentration, as applicable) are required for each spike injection.
8.2.7.2.4 Spike Dilution Factor (DF). For each spike injection, DF, the dilution factor must be determined. DF is the ratio of the total volumetric flow rate of gas through the measurement system to the spike gas flow rate. This factor must be ≥5. The spiking mass balance calculation is directly dependent on the accuracy of the DF determination. As a result, high accuracy total volumetric flow rate and spike gas flowrate measurements are required. These flow rates may be determined by direct or indirect measurement. Calibrated flow meters, venturies, orifices or tracer gas measurements are examples of potential flow measurement techniques.
8.2.7.2.5 Concentrations. The measurement system must record total vapor phase Hg concentrations continuously during the dynamic spiking procedure. It is possible that dynamic spiking at a level close to 200 percent of the native Hg concentration may cause the measured Hg concentration to exceed the calibration span value. Avoid this by choosing a lower spiking level or by recalibration at a higher span. The measurements shall not exceed 120 percent of the calibration span. The “baseline” measurements made between spikes may represent the native Hg concentration (if spike gas flow is stopped between injections) or the native Hg concentration diluted by blank or carrier gas flowing at the same rate as the spike gas (if gas flow cannot be stopped between injections). Each baseline measurement must include at least 4 readings or 1 minute (whichever is greater) of stable responses. Use Equation 30A-10 or 30A-11 in Section 12.10 (as applicable) to convert baseline measurements to native concentration.
8.2.7.2.6 Recovery. Calculate spike recoveries using Equation 30A-7 in Section 12.7. Mass recoveries may be calculated from stable responses based on injected mass flows or from integrated response peaks based on total mass injected. Calculate the mean and RSD for the three (or more) spike injections and compare to the specifications in Section 13.5.
8.2.7.2.7 Error Adjustment Option. You may adjust the measurement data collected during dynamic spiking for the system calibration error using Equation 30A-3 in Section 12. To do this, perform the initial system integrity check prior to the dynamic spiking test, and perform another system integrity check following the dynamic spiking test and before the first test run. If you choose this option, you must apply Equation 30A-3 to both the spiked sample concentration measurement (Css) and the baseline or native concentration measurement (Cnative), each substituted in place of Cavg in the equation.
8.2.7.3 Example Spiking Procedure Using a Hot Vapor Calibration Source Generator.
(a) Introduce the spike gas into the probe using a hot vapor calibration source generator and a solution of HgCl2 in dilute HC1 and HNO3. The calibrator uses a mass flow controller (accurate within 2 percent) to measure the gas flow, and the solution feed is measured using a top-loading balance accurate to 0.01g. The challenges of injecting oxidized Hg may make it impractical to stop the flow of gas between spike injections. In this case, operate the hot vapor calibration source generator continuously during the spiking procedure, swapping blank solutions for HgCl2 solutions when switching between spiking and baseline measurements.
(b) If applicable, monitor the measurement system to make sure the total sampling system flow rate and the sample dilution ratio do not change during this procedure. Record all data on a data sheet similar to Table 30A-5 in Section 17.0. If the Hg measurement system design makes it impractical to measure the total volumetric flow rate through the system, use a spike gas that includes a tracer for measuring the dilution factor, DF (see Equation 30A-9 in Section 12.9). Allow the measurements to stabilize between each spike injection, average the pre- and post-injection baseline measurements, and calculate the native concentration. If this measurement shifts by more than 5 percent during any injection, it may be necessary to discard that data point and repeat the injection to achieve the required RSD among the injections. If the spikes persistently show poor repeatability, or if the recoveries are not within the range specified in Section 13.5, take corrective action.
8.2.8 Run Validation. How do I confirm that each run I conduct is valid?
8.2.8.1 System Integrity Checks.
(a) Before and after each test run, perform a two-point system integrity check using the same procedure as the initial system integrity check described in Section 8.2.5. You may use data from that initial system integrity Start Printed Page 51506check as the pre-run data for the first test run, provided it is the most recent system integrity check done before the first run. You may also use the results of a successful post-run system integrity check as the pre-run data for the next test run. Do not make any adjustments to the measurement system during these checks, other than to maintain the target calibration gas flow rate and the proper dilution ratio.
(b) As a time-saving alternative, you may, at the risk of invalidating multiple test runs, skip one or more integrity checks during a test day. Provided there have been no auto-calibrations or other instrument alterations, a single integrity check may suffice as a post-run check to validate (or invalidate) as many consecutive test runs as can be completed during a single test day. All subsequent test days must begin with a pre-run system integrity check subject to the same performance criteria and corrective action requirements as a post-run system integrity check.
(c) Each system integrity check must meet the criteria for system integrity checks in Section 13.2. If a post-run system integrity check is failed, all test runs since the last passed system integrity check are invalid. If a post-run or a pre-run system integrity check is failed, you must take corrective action and pass another 3-point Hg0 system calibration error test (Section 8.2.4) followed by another system integrity check before conducting any additional test runs. Record the results of the pre- and post-run system integrity checks on a form similar to Table 30A-2 in Section 17.0.
8.2.8.2 Drift Check. Using the data from the successful pre- and post-run system integrity checks, calculate the zero and upscale drift, using Equation 30A-2 in Section 12.3. Exceeding the Section 13.3 specification does not invalidate the run, but corrective action must be taken and a new 3-point Hg0 system calibration error test and a system integrity check must be passed before any more runs are made.
8.3 Dilution-Type Systems—Special Considerations. When a dilution-type measurement system is used, there are three important considerations that must be taken into account to ensure the quality of the emissions data. First, the critical orifice size and dilution ratio must be selected properly so that the sample dew point will be below the sample line and analyzer temperatures. Second, a high-quality, accurate dilution controller must be used to maintain the correct dilution ratio during sampling. The dilution controller should be capable of monitoring the dilution air pressure, orifice upstream pressure, eductor vacuum, and sample flow rates. Third, differences between the molecular weight of calibration gas mixtures, dilution air, and the stack gas molecular weight must be considered because these can affect the dilution ratio and introduce measurement bias.
8.4 Sampling.
(a) Position the probe at the first sampling point. Allow the system to flush and equilibrate for at least two times the measurement system response time before recording any data. Then, traverse and record measurements at all required sampling points. Sample at each traverse point for an equal length of time, maintaining the appropriate sample flow rate or dilution ratio (as applicable). For all Hg instrumental method systems, the minimum sampling time at each sampling point must be at least two times the system response time, but not less than 10 minutes. For concentrating systems, the minimum sampling time must also include at least 4 concentration measurement cycles.
(b) After recording data for the appropriate period of time at the first traverse point, you may move the sample probe to the next point and continue recording, omitting the requirement to allow the system to equilibrate for two times the system response time before recording data at the subsequent traverse points. You must, however, sample at this and all subsequent traverse points for the required minimum amount of time specified in this section. If you must remove the probe from the stack for any reason, you must again allow the sampling system to equilibrate for at least two times the system response time prior to resuming data recording.
(c) If at any point the measured Hg concentration exceeds the calibration span value, you must at a minimum identify and report this as a deviation from the method. Depending on the data quality objectives of the test, this event may require corrective action before proceeding. If the average Hg concentration for any run exceeds the calibration span value, the run is invalidated.
8.5 Moisture Correction. If the moisture basis (wet or dry) of the measurements made with this method is different from the moisture basis of either: (1) The applicable emission limit; or (2) a Hg CEMS or sorbent trap monitoring system being evaluated for relative accuracy, you must determine the moisture content of the flue gas and correct the measured gas concentrations to a dry basis using Method 4 in appendix A-3 of this part or other appropriate methods, subject to the approval of the Administrator.
8.6 Optional Interference Test Procedure.
(a) Select an appropriate calibration span that reflects the source(s) to be tested and perform the interference check at 40 percent of the lowest calibration span value anticipated, e.g., 10 μg/m3. Alternatively, successfully conducting the interference test at an absolute Hg concentration of 2 μg/m3 will demonstrate performance for an equivalent calibration span of 5 μg/m3, the lowest calibration span allowed for Method 30A testing. Therefore, performing the interference test at the 2 μ/m3 level will serve to demonstrate acceptable performance for all calibration spans greater than or equal to 5 μg/m3.
(b) Introduce the interference test gases listed in Table 30A-3 in Section 17.0 into the measurement system separately or as a mixture. The interference test gases HCl and NO must be introduced as a mixture. The interference test gases must be introduced into the sampling system at the probe such that the interference gas mixtures pass through all filters, scrubbers, conditioners, and other components as would be configured for normal sampling.
(c) The interference test must be performed using HgCl2, and each interference test gas (or gas mixture) must be evaluated in triplicate. This is accomplished by measuring the Hg response first with only the HgCl2 gas present and then when adding the interference test gas(es) while maintaining the HgCl2 concentration of the test stream constant. It is important that the equipment used to conduct the interference test be of sufficient quality so as to be capable of blending the HgCl2 and interference gases while maintaining the Hg concentration constant. Gas blending system or manifolds may be used.
(d) The duration of each test should be for a sufficient period of time to ensure the Hg measurement system surfaces are conditioned and a stable output is obtained. Measure the Hg response of the analyzer to these gases in μg/m3. Record the responses and determine the overall interference response using Table 30A-4 in Section 17.0 and the equations presented in Section 12.11. The specification in Section 13.4 must be met.
(e) A copy of these data, including the date completed and a signed certification, must be included with each test report. The intent of this test is that the interference test results are intended to be valid for the life of the system. As a result, the Hg measurement system should be operated and tested in a configuration consistent with the configuration that will be used for field applications. However, if the system used for field testing is not consistent with the system that was interference-tested, the interference test must be repeated before it is used for any field applications. Examples of such conditions include, but are not limited to: major changes in dilution ratio (for dilution based systems), changes in catalyst materials, changes in filtering device design or materials, changes in probe design or configuration, and changes in gas conditioning materials or approaches.
9.0 Quality Control
What quality control measures must I take?
The table which follows is a summary of the mandatory, suggested, and alternative quality assurance and quality control measures and the associated frequency and acceptance criteria. All of the QC data, along with the run data, must be documented and included in the test report. Start Printed Page 51507
Summary Table of QA/QC Requirements
Status 1 Process or element QA/QC specification Acceptance criteria Checking frequency S Identify Data User Regulatory Agency or other primary end user of data Before designing test. M Analyzer Design Analyzer range Sufficiently > high-level gas to allow determination of system calibration error S Analyzer resolution or sensitivity < 2.0 % of full-scale range Manufacturer design. S Interference response Overall response ≤ 3% of calibration span Alternatively, overall response ≤ 0.3 μg/m3 M Calibration Gases Traceability protocol Validation of concentration required M High-level Hg0 gas Equal to the calibration span Each calibration error test. M Mid-level Hg0 gas 40 to 60% of calibration span Each calibration error test. M Low-level Hg0 gas 10 to 30% of calibration span Each calibration error test. M High-level HgCl2 gas Equal to the calibration span Each system integrity check (if it better represents Cnative than the mid level gas). M Mid-level HgCl2 40 to 60% of calibration span Each system gas integrity check (if it better represents Cnative than the high level gas). M Zero gas Each system integrity check. M Dynamic spike gas (Cnative ≥ 1 μg/m3) A high-concentration HgCl2 gas, used to produce a spiked sample concentration that is 150 to 200% of the native concentration Pre-test; dynamic spiking not required until 1/1/09. M Dynamic spike gas (Cnative < 1 μg/m3) A high-concentration HgCl2 gas, used to produce a spiked sample concentration that is 1 to 2 μg/m3 above the native concentration Pre-test; dynamic spiking not required until 1/1/09. S Data Recorder Design Data resolution ≤ 0.5% of full-scale Manufacturer design. M Sample Extraction Probe material Inert to sample constituents (e.g., PFA Teflon, or quartz if stack > 500 °F) Each run. M Sample Extraction Probe, filter and sample line temperature For dry-basis analyzers, keep sample above the dew point, by heating prior to moisture removal For wet-basis analyzers, keep sample above dew point at all times, by heating or dilution Each run. M Sample Extraction Calibration valve material Inert to sample constituents (e.g., PFA Teflon or PFA Teflon coated) Each test. S Sample Extraction Sample pump material Inert to sample constituents Each test. M Sample Extraction Manifold material Inert to sample constituents Each test. M Particulate Removal Filter inertness Pass calibration error check Each calibration error check. M System Calibration Performance System calibration error (CE) test CE ≤ 5.0 % of the calibration span for the low-, mid-or high-level Hg0 calibration gas Alternative specification: ≤ 0.5 μg/m3 absolute difference between system response and reference value Before initial run and after a failed system integrity check or drift test. M System Calibration Performance System integrity check Error ≤ 5.0% of the calibration span for the zero and mid- or high-level HgCl2 calibration gas Alternative specification: ≤ 0.5 μg/m3 absolute difference between system response and reference value Before initial run, after each run, at the beginning of subsequent test days, and after a failed system integrity check or drift test. M System Performance System response time Used to determine minimum sampling time per point During initial 3-point system calibration error test. M System Performance Drift ≤ 3.0% of calibration span for the zero and mid- or high-level gas Alternative specification: ≤ 0.3 μg/m3 absolute difference between pre- and post-run system calibration error percentages. At least once per test day. M System Performance Minimum sampling time The greater of two times the system response time or 10 minutes. Concentrating systems must also include at least 4 cycles Each sampling point. M System Performance Percentage spike recovery and relative standard deviation Percentage spike recovery, at the target level: 100 ± 10% Relative standard deviation: ≤ 5 percent Alternative specification: absolute difference between calculated and measured spike values ≤ 0.5 μg/m3 Before initial dynamic spiking not required until 1/1/09. Start Printed Page 51508 M Sample Point Selection Number and Location of Sample Points For emission testing applications, use 12 points, located according to Method 1 in appendix A-1 to this part, unless the results of a stratification test allow fewer points to be used Prior to first run. For Part 60 RATAs, follow the procedures in Performance Specification 2, section 8.1.3, and for Part 75 RATAs, follow the procedures in section 6.5.6 of appendix A to Part 75. That is: • At any test location, you may use 3 sample points located at 16.7, 50.0, and 83.3% of a “long” measurement line passing through the centroidal area; or • At any test location, you may use 6 sample points along a diameter, located according to Method 1 (Part 75 RATAs, only); or • At a location where stratification is not expected and the measurement line is > 2.4 m (7.8 ft), you may use 3 sample points located along a “short” measurement line at 0.4, 1.0, and 2.0 m from the stack or duct wall or, for Part 75 only, sample points may be located at 4.4, 14.6, and 29.6% of the measurement line; or • After a wet scrubber or at a point where dissimilar gas streams are combined, either locate 3 sample points along the “long” measurement line or locate 6 Method 1 points along a diameter (Part 75, only), unless the results of a stratification test allow you to use a “short” 3-point measurement line or to sample at a single point • If it can be demonstrated that stack gas concentration is ≤ 3 μg/m3, then the test site is exempted from stratification testing. Use the 3-point “short” measurement line if the stack diameter is > 2.4 m (7.8 ft) and the 3-point “long” line for stack diameters ≤ 2.4 m (7.8 ft) A Sample Point Selection Stratification Test (see Section 8.1.3) If the Hg concentration 2 at each traverse point during the stratification test is: • Within ± 5% of mean, use 1-point sampling (at the point closest to the mean); or • Not within ± 5% of mean, but is within ± 10% of mean, use 3-point sampling. Locate points according to Section 8.1.3.2.2 of this method Prior to first run. Alternatively, if the Hg concentration at each point is: • Within ± 0.2 μg/m3 of mean, use 1-point sampling (at the point closest to the mean); or • Not within ± 0.2 μg/m3 of mean, use 3-point sampling. Locate points according to Section 8.1.3.2.2 of this method Prior to 1/1/09, you may (1) forgo stratification testing and use 3 sampling points (as per Section 8.1.3.2.2) or (2) perform a SO2 stratification test (see Sections 6.5.6.1 and 6.5.6.3 of appendix A to part 75), in lieu of a Hg stratification test. If the test location is unstratified or minimally stratified for SO2, it can be considered unstratified or minimally stratified for Hg also. Start Printed Page 51509 If the Hg concentration is > 10% of the mean at any point, then, if the alternative specification is not met or if the stack diameter is ≤ 2.4 m (7.8 ft): • Perform sampling at 12 Method 1 points; or • Sample at 3 points located at 16.7, 50.0 and 83.3% of the measurement line that exhibited the highest average Hg concentration during stratification test; or • Sample at 6 Method 1 points along the line that exhibited the highest average Hg concentration (Part 75 RATAs, only) On and after 1/1/09, only Hg stratification tests are acceptable for the purposes of this method. M Data Recording Frequency Once per cycle During run. S Data Parameters Sample concentration and calibration span All analyzer readings during each run within calibration span Each run. M Data Parameters Sample concentration and calibration span All analyzer readings during dynamic spiking tests within 120% of calibration span Each spike injection. M Data Parameters Sample concentration and calibration span Average Hg concentration for the run ≤ calibration span Each run. 1 M = Mandatory; S = Suggested; A = Alternative. 2 These may either be the unadjusted Hg concentrations or concentrations normalized to account for temporal variations. 10.0 Calibration and Standardization
What measurement system calibrations are required?
Your analyzer must be calibrated with Hg° standards. The initial 3-point system calibration error test described in Section 8.2.4 is required before you start the test. Also, prior to and following test runs, the two-point system integrity checks described in Sections 8.2.5 and 8.2.8 are required. On and after January 1, 2009, the pre-test dynamic spiking procedure described in section 8.2.7 is also required to verify that the accuracy of the measurement system is suitable and not adversely affected by the flue gas matrix.
11.0 Analytical Procedures
Because sample collection and analysis are performed together (see Section 8), additional discussion of the analytical procedure is not necessary.
12.0 Calculations and Data Analysis
You must follow the procedures for calculations and data analysis listed in this section.
12.1 Nomenclature. The terms used in the equations are defined as follows:
Bws = Moisture content of sample gas as measured by Method 4 in Appendix A-3 to this part, percent/100.
Cavg = Average unadjusted Hg concentration for the test run, as indicated by the data recorder μg/m3.
Cbaseline = Average Hg concentration measured before and after dynamic spiking injections, μg/m3.
Cd = Hg concentration, dry basis, μg/m3.
Cdif = Absolute value of the difference between the measured Hg concentrations of the reference HgCl2 calibration gas, with and without the individual or combined interference gases, μg/m3.
Cdif avg = Average of the 3 absolute values of the difference between the measured Hg concentrations of the reference HgCl2 calibration gas, with and without the individual or combined interference gases, μg/m3.
Cgas = Average Hg concentration in the effluent gas for the test run, adjusted for system calibration error, μg/m3.
Cint = Measured Hg concentration of the reference HgCl2 calibration gas plus the individual or combined interference gases, μg/m3.
Cm = Average of pre- and post-run system integrity check responses for the upscale (i.e., mid- or high-level) calibration gas, μg/m3.
Cma = Actual concentration of the upscale (i.e., mid- or high-level) calibration gas used for the system integrity checks, μg/m3.
C0 = Average of pre- and post-run system integrity check responses from the zero gas, μg/m3.
Cnative = Vapor phase Hg concentration in the source effluent, μg/m3.
Cref = Measured Hg concentration of the reference HgCl2 calibration gas alone, in the interference test, μg/m3.
Cs = Measured concentration of a calibration gas (zero-, low-, mid-, or high-level), when introduced in system calibration mode, μg/m3.
Cspike = Actual Hg concentration of the spike gas, μg/m3.
C*spike = Hg concentration of the spike gas required to achieve a certain target value for the spiked sample Hg concentration, μg/m3.
Css = Measured Hg concentration of the spiked sample at the target level, μg/m3.
C*ss = Expected Hg concentration of the spiked sample at the target level, μg/m3.
Ctarget = Target Hg concentration of the spiked sample, μg/m3.
CTnative = Measured tracer gas concentration present in native effluent gas, ppm.
CTdir = Tracer gas concentration injected with spike gas, ppm.
CTv = Diluted tracer gas concentration measured in a spiked sample, ppm.
Cv = Certified Hg° or HgCl2 concentration of a calibration gas (zero, low, mid, or high), μg/m3.
Cw = Hg concentration measured under moist sample conditions, wet basis, μg/m3.
CS = Calibration span, μg/m3.
D = Zero or upscale drift, percent of calibration span.
DF = Dilution factor of the spike gas, dimensionless.
I = Interference response, percent of calibration span.
Qprobe = Total flow rate of the stack gas sample plus the spike gas, liters/min.
Qspike = Flow rate of the spike gas, liters/min.
Ri = Individual injection spike recovery, %;.
R= Mean value of spike recoveries at a particular target level, %;.
RSD = Relative standard deviation, %;.
SCE = System calibration error, percent of calibration span.
SCEi = Pre-run system calibration error during the two-point system integrity check, percent of calibration span.
SCEf = Post-run system calibration error during the two-point system integrity check, percent of calibration span.
12.2 System Calibration Error. Use Equation 30A-1 to calculate the system calibration error. Equation 30A-1 applies to: 3-point system calibration error tests performed with Hg° standards; and pre- and post-run two-point system integrity checks performed with HgCl2.

12.3 Drift Assessment. Use Equation 30A-2 to separately calculate the zero and upscale drift for each test run.

12.3 Effluent Hg Concentration. For each test run, calculate Cavg, the arithmetic average of all valid Hg concentration values recorded during the run. Then, adjust the value of CavgStart Printed Page 51510for system calibration error, using Equation 30A-3.

12.4 Moisture Correction. Use Equation 30A-4a if your measurements need to be corrected to a dry basis.

Use Equation 30A-4b if your measurements need to be corrected to a wet basis.

12.5 Dynamic Spike Gas Concentrations. Use Equation 30A-5 to determine the spike gas concentration needed to produce a spiked sample with a certain “target” Hg concentration.

12.6 Spiked Sample Concentration. Use Equation 30A-6 to determine the expected or theoretical Hg concentration of a spiked sample.

12.7 Spike Recovery. Use Equation 30A-7 to calculate the percentage recovery of each spike.

12.8 Relative Standard Deviation. Use Equation 30A-8 to calculate the relative standard deviation of the individual percentage spike recovery values from the mean.

12.9 Spike Dilution Factor. Use Equation 30A-9 to calculate the spike dilution factor, using either direct flow measurements or tracer gas measurements.

12.10 Native Concentration. For spiking procedures that inject blank or carrier gases (at the spiking flow rate, Qspike) between spikes, use Equation 30A-10 to calculate the native concentration.

For spiking procedures that halt all injections between spikes, the native concentration equals the average baseline concentration (see Equation 30A-11).

12.11 Overall Interference Response. Use equation 30A-12 to calculate the overall interference response.

Where, for each interference gas (or mixture):

Start Printed Page 51511
13.0 Method Performance
13.1 System Calibration Error Test. This specification applies to the 3-point system calibration error tests using Hg0. At each calibration gas level tested (low-, mid-, or high-level), the calibration error must be within ±5.0 percent of the calibration span. Alternatively, the results are acceptable if | Cs − Cv | ≤0.5 μg/m3.
13.2 System Integrity Checks. This specification applies to all pre- and post-run 2-point system integrity checks using HgCl2 and zero gas. At each calibration gas level tested (zero and mid- or high-level), the error must be within ±5.0 percent of the calibration span. Alternatively, the results are acceptable if | Cs − Cv | ≤0.5 μg/m3.
13.3 Drift. For each run, the low-level and upscale drift must be less than or equal to 3.0 percent of the calibration span. The drift is also acceptable if the pre- and post-run system integrity check responses do not differ by more than 0.3 μg/m3 (i.e., | Cs post-run − Cs pre-run | ≤0.3 μg/m3).
13.4 Interference Test. Summarize the results following the format contained in Table 30A-4. For each interference gas (or mixture), calculate the mean difference between the measurement system responses with and without the interference test gas(es). The overall interference response for the analyzer that was used for the test (calculated according to Equation 30A-12), must not be greater than 3.0 percent of the calibration span used for the test (see Section 8.6). The results of the interference test are also acceptable if the sum of the absolute average differences for all interference gases (i.e., Σ Cdif avg) does not exceed 0.3 μg/m3.
13.5 Dynamic Spiking Test. For the pre-test dynamic spiking, the mean value of the percentage spike recovery must be 100 ±10 percent. In addition, the relative standard deviation (RSD) of the individual percentage spike recovery values from the mean must be ≤5.0 percent. Alternatively, if the mean percentage recovery is not met, the results are acceptable if the absolute difference between the theoretical spiked sample concentration (see Section 12.6) and the actual average value of the spiked sample concentration is ≤0.5 μg/m3.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. EPA Traceability Protocol for Qualification and Certification of Elemental Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
2. EPA Traceability Protocol for Qualification and Certification of Oxidized Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
3. EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards, expected revision publication date December 2008, see www.epa.gov/ttn/emc.
17.0 Figures and Tables
Start Printed Page 51512
Start Printed Page 51513
Start Printed Page 51514
Start Printed Page 51515
 Start Printed Page 51516
Start Printed Page 51516Table 30A-3.—Interference Check Gas Concentrations
Potential interferent gas 1 Concentration, tentative—(balance N2) CO2 15% ± 1% CO2 CO 100 ± 20 ppm HCl 2 100 ± 20 ppm NO 2 250 ± 50 ppm SO2 200 ± 20 ppm O2 3% ± 1% O2 H2 O 10% ± 1% H2 O Nitrogen Balance Other 1 Any of these specific gases can be tested at a lower level if the manufacturer has provided reliable means for limiting or scrubbing that gas to a specified level. 2 HCl and NO must be tested as a mixture.
Start Printed Page 51517
Start Printed Page 51518
Method 30B—Determination of Total Vapor Phase Mercury Emissions From Coal-Fired Combustion Sources Using Carbon Sorbent Traps
1.0 Scope and Application
What is Method 30B?
Method 30B is a procedure for measuring total vapor phase mercury (Hg) emissions from coal-fired combustion sources using sorbent trap sampling and an extractive or thermal analytical technique. This method is only intended for use only under relatively low particulate conditions (e.g., sampling after all pollution control devices). Quality assurance and quality control requirements are included to assure that you, the tester, collect data of known and acceptable quality for each testing program. This method does not completely describe all equipment, supplies, and sampling and analytical procedures you will need, but instead refers to other test methods for some of the details. Therefore, to obtain reliable results, you should also have a thorough knowledge of these additional methods which are found in Appendices A-1 and A-3 to this part:
(a) Method 1—Sample and Velocity Traverses for Stationary Sources.
(b) Method 4—Determination of Moisture Content in Stack Gases.
(c) Method 5—Determination of Particulate Matter Emissions from Stationary Sources
1.1 Analytes. What does this method determine? This method is designed to measure the mass concentration of total vapor phase Hg in flue gas, including elemental Hg (Hg0) and oxidized forms of Hg (Hg+2), in micrograms per dry standard cubic meter (μg/dscm).
Analyte CAS No. Analytical range and sensitivity Elemental Hg (Hg 0 ) 7439-97-6 Typically 0.1 μg/dscm to >50 μg/dscm. Oxidized Hg (Hg+2) (Same) 1.2 Applicability. When is this method required? Method 30B is a reference method for relative accuracy test audits (RATAs) of vapor phase Hg CEMS and sorbent trap monitoring systems installed at coal-fired boilers and is also appropriate for Hg emissions testing at such boilers. It is intended for use only under relatively low particulate conditions (i.e., sampling after all pollution control devices); in cases where significant amounts of particle-bound Hg may be present, an isokinetic sampling method for Hg should be used. Method 30B may also be specified by New Source Performance Standards (NSPS), National Emission Standards for Hazardous Air Pollutants (NESHAP), emissions trading programs, State Implementation Plans (SIPs), and operating permits that require measurement of Hg concentrations in stationary source emissions, either to determine compliance with an applicable emission standard or limit, or to conduct RATAs of Hg CEMS and sorbent trap monitoring systems.
1.3 Data Quality Objectives (DQO). How good must my collected data be? Method 30B has been designed to provide data of high and known quality for Hg emissions testing and for RATA testing of Hg monitoring systems, including CEMS and sorbent trap monitors. In these and other applications, the principal objective is to ensure the accuracy of the data at the actual emissions levels and in the actual emissions matrix encountered. To meet this objective, NIST-traceable calibration standards must be used and method performance tests are required.
2.0 Summary of Method
Known volumes of flue gas are extracted from a stack or duct through paired, in-stack sorbent media traps at an appropriate flow rate. Collection of mercury on the sorbent media in the stack mitigates potential loss of mercury during transport through a probe/sample line. For each test run, paired train sampling is required to determine measurement precision and verify acceptability of the measured emissions data. A field recovery test which assesses recovery of an elemental Hg spike to determine measurement bias is also used to verify data acceptability. The sorbent traps are recovered from the sampling system, prepared for analysis as needed, and analyzed by any suitable determinative technique that can meet the performance criteria.
3.0 Definitions
3.1 Analytical System is the combined equipment and apparatus used to perform sample analyses. This includes any associated sample preparation apparatus e.g., digestion equipment, spiking systems, reduction devices, etc., as well as analytical instrumentation such as UV AA and UV AF cold vapor analyzers.
3.2 Calibration Standards are the Hg containing solutions prepared from NIST traceable standards and are used to directly calibrate analytical systems.
3.3 Independent Calibration Standard is a NIST traceable standard obtained from a source or supplier independent of that for the calibration standards and is used to confirm the integrity of the calibration standards used.
3.4 Method Detection Limit (MDL) is the lowest mass of Hg greater than zero that can be estimated and reported by your candidate analytical technique. The MDL is statistically derived from replicate low level measurements near your analytical instrument's detection level.
3.5 NIST means the National Institute of Standards and Technology, located in Gaithersburg, Maryland.
3.6 Run means a series of gas samples taken successively from the stack or duct. A test normally consists of a specific number of runs.
3.7 Sorbent Trap means a cartridge or sleeve containing a sorbent media (typically activated carbon treated with iodine or some other halogen) with multiple sections separated by an inert material such as glass wool. These sorbent traps are optimized for the quantitative capture of elemental and oxidized forms of Hg and can be analyzed by multiple techniques.
3.8 Test refers to the series of runs required by the applicable regulation.
3.9 Thermal Analysis means an analytical technique where the contents of the sorbent traps are analyzed using a thermal technique (desorption or combustion) to release the captured Hg in a detectable form for quantification.
3.10 Wet Analysis means an analytical technique where the contents of the sorbent tube are first leached or digested to quantitatively transfer the captured Hg to liquid solution for subsequent analysis.
4.0 Interferences
Interferences may result from the sorbent trap material used as well as from the measurement environment itself. The iodine present on some sorbent traps may impart a negative measurement bias. High levels of sulfur trioxide (SO3) are also suspected to compromise the performance of sorbent trap Hg capture. These, and other, potential interferences are assessed by performing the analytical matrix interference, Hg0 and HgCl2 analytical bias and field recovery tests.
5.0 Safety
What safety measures should I consider when using this method? This method may require you to work with hazardous materials and in hazardous conditions. You are encouraged to establish safety procedures before using the method. Among other precautions, you should become familiar with the safety recommendations in the gas analyzer user's manual. Occupational Safety and Health Administration (OSHA) regulations concerning use of compressed gas cylinders and noxious gases may apply.
5.1 Site Hazards. Prior to applying these procedures/specifications in the field, the potential hazards at the test site should be considered; advance coordination with the site is critical to understand the conditions and applicable safety policies. At a minimum, portions of the sampling system will be hot, requiring appropriate gloves, long sleeves, and caution in handling this equipment.
5.2 Laboratory Safety. Policies should be in place to minimize risk of chemical exposure and to properly handle waste disposal in the laboratory. Personnel shall wear appropriate laboratory attire according to a Chemical Hygiene Plan established by the laboratory.
5.3 Reagent Toxicity/Carcinogenicity. The toxicity and carcinogenicity of any reagents used must be considered. Depending upon the sampling and analytical technologies selected, this measurement may involve hazardous materials, operations, and equipment and this method does not address all of the safety problems associated with implementing this approach. It is the responsibility of the user to establish appropriate safety and health practices and determine the applicable regulatory limitations prior to performance. Any chemical should be regarded as a potential health hazard and exposure to these compounds should be minimized. Chemists should refer to the Material Safety Data Sheet (MSDS) for each chemical used.
5.4 Waste Disposal. Any waste generated by this procedure must be disposed of Start Printed Page 51519according to a hazardous materials management plan that details and tracks various waste streams and disposal procedures.
6.0 Equipment and Supplies
The following list is presented as an example of key equipment and supplies likely required to measure vapor-phase Hg using a sorbent trap sampling system. It is recognized that additional equipment and supplies may be needed. Collection of paired samples is required.
6.1 Sorbent Trap Sampling System. A typical sorbent trap sampling system is shown in Figure 30B-1 in Section 17.0. The sorbent trap sampling system shall include the following components:
6.1.1 Sorbent Traps. The sorbent media used to collect Hg must be configured in a trap with at least two distinct segments or sections, connected in series, that are amenable to separate analyses. Section 1 is designated for primary capture of gaseous Hg. Section 2 is designated as a backup section for determination of vapor phase Hg breakthrough. Each sorbent trap must be inscribed or otherwise permanently marked with a unique identification number, for tracking purposes. The sorbent media may be any collection material (e.g., carbon, chemically-treated filter, etc.) capable of quantitatively capturing and recovering for subsequent analysis, all gaseous forms of Hg in the emissions from the intended application. Selection of the sorbent media shall be based on the material's ability to achieve the performance criteria contained in this method as well as the sorbent's vapor phase Hg capture efficiency for the emissions matrix and the expected sampling duration at the test site. The sorbent media must be obtained from a source that can demonstrate their quality assurance and quality control (see Section 7.2). The paired sorbent traps are supported on a probe (or probes) and inserted directly into the flue gas stream.
6.1.2 Sampling Probe Assembly. Each probe assembly shall have a leak-free attachment to the sorbent trap(s). Each sorbent trap must be mounted at the entrance of or within the probe such that the gas sampled enters the trap directly. Each probe/sorbent trap assembly must be heated to a temperature sufficient to prevent liquid condensation in the sorbent trap(s). Auxiliary heating is required only where the stack temperature is too low to prevent condensation. Use a calibrated thermocouple to monitor the stack temperature. A single probe capable of operating the paired sorbent traps may be used. Alternatively, individual probe/sorbent trap assemblies may be used, provided that the individual sorbent traps are co-located to ensure representative Hg monitoring.
6.1.3 Moisture Removal Device. A moisture removal device or system shall be used to remove water vapor from the gas stream prior to entering dry gas flow metering devices.
6.1.4 Vacuum Pump. Use a leak-tight, vacuum pump capable of operating within the system's flow range.
6.1.5 Gas Flow Meter. A gas flow meter (such as a dry gas meter, thermal mass flow meter, or other suitable measurement device) shall be used to determine the total sample volume on a dry basis, in units of standard cubic meters. The meter must be sufficiently accurate to measure the total sample volume to within 2 percent and must be calibrated at selected flow rates across the range of sample flow rates at which the sampling train will be operated. The gas flow meter shall be equipped with any necessary auxiliary measurement devices (e.g., temperature sensors, pressure measurement devices) needed to correct the sample volume to standard conditions.
6.1.6 Sample Flow Rate Meter and Controller. Use a flow rate indicator and controller for maintaining necessary sampling flow rates.
6.1.7 Temperature Sensor. Same as Section 6.1.1.7 of Method 5 in Appendix A-3 to this part.
6.1.8 Barometer. Same as Section 6.1.2 of Method 5 in Appendix A-3 to this part.
6.1.9 Data Logger (optional). Device for recording associated and necessary ancillary information (e.g., temperatures, pressures, flow, time, etc.).
6.2 Gaseous Hg0 Sorbent Trap Spiking System. A known mass of gaseous Hg0 must be either present on or spiked onto the first section of sorbent traps in order to perform the Hg0 and HgCl2 analytical bias test and the field recovery study. Any approach capable of quantitatively delivering known masses of Hg0 onto sorbent traps is acceptable. Several spiking technologies or devices are available to meet this objective. Their practicality is a function of Hg mass spike levels. For low levels, NIST-certified or NIST-traceable gas generators or tanks may be suitable. An alternative system, capable of delivering almost any mass required, makes use of NIST-certified or NIST-traceable Hg salt solutions (e.g., HgCl2, Hg(NO3)2). With this system, an aliquot of known volume and concentration is added to a reaction vessel containing a reducing agent (e.g., stannous chloride); the Hg salt solution is reduced to Hg0 and purged onto the sorbent trap using an impinger sparging system. When available, information on example spiking systems will be posted at http://www.epa.gov/ttn/emc.
6.3 Sample Analysis Equipment. Any analytical system capable of quantitatively recovering and quantifying total Hg from the sorbent media selected is acceptable provided that the analysis can meet the performance criteria described in this method. Example recovery techniques include acid leaching, digestion, and thermal desorption/direct combustion. Example analytical techniques include, but are not limited to, ultraviolet atomic fluorescence (UV AF), ultraviolet atomic absorption (UV AA) with and without gold trapping, and X-ray fluorescence (XRF) analysis.
6.3 Moisture Measurement System. If correction of the measured Hg emissions for moisture is required (see Section 8.3.3.7), either Method 4 in Appendix A-3 to this part or other moisture measurement methods approved by the Administrator will be needed to measure stack gas moisture content.
7.0 Reagents and Standards
7.1 Reagents and Standards. Only NIST-certified or NIST-traceable calibration standards, standard reference materials, and reagents shall be used for the tests and procedures required by this method.
7.2 Sorbent Trap Media. The sorbent trap media shall be prepared such that the material used for testing is of known and acceptable quality. Sorbent supplier quality assurance/quality control measures to ensure appropriate and consistent performance such as sorptive capacity, uniformity of preparation treatments, and background levels shall be considered.
8.0 Sample Collection and Handling
This section presents the sample collection and handling procedures along with the pretest and on-site performance tests required by this method. Since you may choose different options to comply with certain performance criteria, each test report must identify the specific options selected and document the results with respect to the performance criteria of this method.
8.1 Sample Point Selection. What sampling site and sampling points do I select? Same as Section 8.1 of Method 30A of this appendix.
8.2 Measurement System Performance Tests. What performance criteria must my measurement system meet? The following laboratory and field procedures and associated criteria of this section are designed to ensure (1) selection of a sorbent and analytical technique combination capable of quantitative collection and analysis of gaseous Hg, (2) collection of an adequate amount of Hg on each sorbent trap during field tests, and (3) adequate performance of the method for each test program: The primary objectives of these performance tests are to characterize and verify the performance of your intended analytical system and associated sampling and analytical procedures, and to define the minimum amount of Hg (as the sample collection target) that can be quantified reliably.
(a) Analytical Matrix Interference Test;
(b) Determination of Minimum Sample Mass;
(c) Hg0 and HgCl2 Analytical Bias Test;
(d) Determination of Nominal Sample Volume;
(e) Field Recovery Test.
8.2.1 Analytical Matrix Interference Test and Minimum Sample Dilution.
(a) The analytical matrix interference test is a laboratory procedure. It is required only if you elect to use a liquid digestion analytical approach and needs to be performed only once for each sorbent material used. The purpose of the test is to verify the presence or absence of known and potential analytical matrix interferences, including the potential negative bias associated with iodine common to many sorbent trap materials. The analytical matrix interference test determines the minimum dilution (if any) necessary to mitigate matrix effects on the sample digestate solutions.
(b) The result of the analytical matrix interference test, i.e., the minimum sample dilution required (if any) for all sample Start Printed Page 51520analyses, is used to establish the minimum sample mass needed for the Hg0 and HgCl2 analytical bias test and to determine the nominal sample volume for a test run. The analytical matrix interference test is sorbent material-specific and shall be performed for each sorbent material you intend to use for field sampling and analysis. The test shall be performed using a mass of sorbent material comparable to the sorbent mass typically used in the first section of the trap for sampling. Similar sorbent materials from different sources of supply are considered to be different materials and must be tested individually. You must conduct the analytical matrix interference test for each sorbent material prior to the analysis of field samples.
8.2.1.1 Analytical Matrix Interference Test Procedures. Digest and prepare for analysis a representative mass of sorbent material (unsampled) according to your intended laboratory techniques for field samples. Analyze the digestate according to your intended analytical conditions at the least diluted level you intend to use for sample analysis (e.g., undiluted, 1 in 10 dilution, etc.). Determine the Hg concentration of the undiluted digestate solution. Prepare a series of solutions with a fixed final volume containing graduated aliquots of the sample digestate and, a fixed aliquot of a calibration standard (with the balance being Hg-free reagent or H2 0) to establish solutions of varied digestate dilution ratio (e.g., 1:2, 1:5, 1:10, 1:100, etc.—see example in Section 8.2.1.3, below). One of these solutions should contain only the aliquot of the calibration standard in Hg-free reagent or H2 O. This will result in a series of solutions where the amount of Hg is held relatively constant and only the volume of digestate diluted is varied. Analyze each of these solutions following intended sample analytical procedures and conditions, determining the concentration for each solution.
8.2.1.2 Analytical Matrix Interference Test Acceptance Criteria. Compare the measured concentration of each solution containing digestate to the measured concentration of the digestate-free solution. The lowest dilution ratio of any solution having a Hg concentration within ±5 percent of the digestate-free solution is the minimum dilution ratio required for analysis of all samples. If you desire to measure the digestate without dilution, the ± 5 percent criterion must be met at a dilution ratio of at least 9:10 (i.e., ≥90% digestate).
8.2.1.3 Example Analytical Matrix Interference Test. An example analytical matrix interference test is presented below. Additional information on the conduct of the analytical matrix interference test will be posted at http://www.epa.gov/ttn/emc. Determine the most sensitive working range for the analyzer to be used. This will be a narrow range of concentrations. Digest and prepare for analysis a representative mass of sorbent material (unsampled) according to your intended laboratory techniques for sample preparation and analysis. Prepare a calibration curve for the most sensitive analytical region, e.g., 0.0, 0.5, 1.0, 3.0, 5.0, 10 ppb. Using the highest calibration standard, e.g., 10.0 ppb, prepare a series of solutions by adding successively smaller increments of the digestate to a fixed volume of the calibration standard and bringing each solution to a final fixed volume with mercury-free deionized water (diH2 O). To 2.0 ml of the calibration standard add 18.0, 10.0, 4.0, 2.0, 1.0, 0.2, and 0.0 ml of the digestate. Bring the final volume of each solution to a total volume of 20 ml by adding 0.0, 8.0, 14.0, 16.0, 17.0, 17.8, and 18.0 ml of diH2 O. This will yield solutions with dilution ratios of 9:10, 1:2, 1:5, 1:10, 1:20, 1:100, and 0:10, respectively. Determine the Hg concentration of each solution. The dilution ratio of any solution having a concentration that is within ±5 percent of the concentration of the solution containing 0.0 ml of digestate is an acceptable dilution ratio for analyzing field samples. If more than one solution meets this criterion, the one with the lowest dilution ratio is the minimum dilution required for analysis of field samples. If the 9:10 dilution meets this criterion, then no sample dilution is required.
8.2.2 Determination of Minimum Sample Mass. The minimum mass of Hg that must be collected per sample must be determined. This information is necessary in order to effectively perform the Hg0 and HgCl2 Analytical Bias Test, to estimate target sample volumes/sample times for test runs, and to ensure the quality of the measurements. The determination of minimum sample mass is a direct function of analytical technique, measurement sensitivity, dilutions, etc. This determination is required for all analytical techniques. Based on the analytical approach you employ, you should determine the most sensitive calibration range. Based on a calibration point within that range, you must consider all sample treatments (e.g., dilutions) to determine the mass of sample that needs to be collected to ensure that all sample analyses fall within your calibration curve.
8.2.2.1 Determination of Minimum Calibration Concentration or Mass. Based on your instrument's sensitivity and linearity, determine the calibration concentrations or masses that make up a representative low level calibration range. Verify that you are able to meet the multipoint calibration performance criteria in section 11.0 of this method. Select a calibration concentration or mass that is no less than 2 times the lowest concentration or mass in your calibration curve. The lowest point in your calibration curve must be at least 5, and preferably 10, times the Method Detection Limit (MDL), which is the minimum amount of the analyte that can be detected and reported. The MDL must be determined at least once for the analytical system using an MDL study such as that found in section 17.0 of the proposed amendments to EPA Method 301 (69 FR 76642, 12/22/2004).
Note to Section 8.2.2.1: While it might be desirable to base the minimum calibration concentration or mass on the lowest point in the calibration curve, selecting a higher concentration or mass is necessary to ensure that all analyses of the field samples will fall within the calibration curve. Therefore, it is strongly recommended that you select a minimum calibration concentration or mass that is sufficiently above the lowest point of the calibration curve (see examples in sections 8.2.2.2.1 and 8.2.2.2.2 below).
8.2.2.2 Determination of Minimum Sample Mass. Based on your minimum calibration concentration or mass and other sample treatments including, but not limited to, final digestate volume and minimum sample dilution, determine the minimum sample mass. Consideration should also be given to the Hg levels expected to be measured in Section 2 of the sorbent traps and to the breakthrough criteria presented in Table 9-1.
8.2.2.2.1 Example Determination of Minimum Sample Mass for Thermal Desorption Analysis. A thermal analysis system has been calibrated at five Hg mass levels: 10 ng, 20 ng, 50 ng, 100 ng, 200 ng, and shown to meet the calibration performance criteria in this method. Based on 2 times the lowest point in the calibration curve, 20 ng is selected as the minimum calibration mass. Because the entire sample is analyzed and there are no dilutions involved, the minimum sample mass is also 20 ng.
Note: In this example, if the typical background (blank) Hg levels in section 2 were relatively high (e.g., 3 to 5 ng), a sample mass of 20 ng might not have been sufficient to ensure that the breakthrough criteria in Table 9-1 would be met, thereby necessitating the use of a higher point on the calibration curve (e.g., 50 ng) as the minimum calibration and sample mass.
8.2.2.2.2 Example Determination of Minimum Sample Mass for Acid Leachate/Digestate Analysis. A cold vapor analysis system has been calibrated at four Hg concentration levels: 2 ng/L, 5 ng, 10 ng/L, 20 ng/L, and shown to meet the calibration performance criteria in this method. Based on 2 times the lowest point in the calibration curve, 4 ng/L was selected as the minimum calibration concentration. The final sample volume of a digestate is nominally 50 ml (0.05 L) and the minimum dilution necessary was determined to be 1:100 by the Analytical Matrix Interference Test of Section 8.2.1. The following calculation would be used to determine the minimum sample mass.
Minimum sample mass = (4 ng/L) × (0.05 L) × (100) = 20 ng
Note: In this example, if the typical background (blank) Hg levels in section 2 were relatively high (e.g., 3 to 5 ng), a sample mass of 20 ng might not have been sufficient to ensure that the breakthrough criterion in Table 9-1 would be met, thereby necessitating the use of a higher point on the calibration curve (e.g., 10 ng/L) as the minimum calibration concentration.
8.2.3 Hg0 and HgCl2 Analytical Bias Test. Before analyzing any field samples, the laboratory must demonstrate the ability to recover and accurately quantify Hg0 and HgCl2 from the chosen sorbent media by performing the following analytical bias test for sorbent traps spiked with Hg0 and HgCl2. The analytical bias test is performed at a minimum of two distinct sorbent trap Hg loadings that will: (1) Represent the lower and upper bound of sample Hg loadings for application of the analytical technique to the Start Printed Page 51521field samples, and (2) be used for data validation.
8.2.3.1 Hg0 and HgCl2 Analytical Bias Test Procedures. Determine the lower and upper bound mass loadings. The minimum sample mass established in Section 8.2.2.2 can be used for the lower bound Hg mass loading although lower Hg loading levels are acceptable. The upper bound Hg loading level should be an estimate of the greatest mass loading that may result as a function of stack concentration and volume sampled. As previously noted, this test defines the bounds that actual field samples must be within in order to be valid.
8.2.3.1.1 Hg0 Analytical Bias Test. Analyze the front section of three sorbent traps containing Hg0 at the lower bound mass loading level and the front section of three sorbent traps containing Hg0 at the upper bound mass loading level. In other words, analyze each mass loading level in triplicate. You may refer to Section 6.2 for spiking guidance. Prepare and analyze each spiked trap, using the same techniques that will be used to prepare and analyze the field samples. The average recovery for the three traps at each mass loading level must be between 90 and 110 percent. If multiple types of sorbent media are to be analyzed, a separate analytical bias test is required for each sorbent material.
8.2.3.1.2 HgCl2 Analytical Bias Test. Analyze the front section of three sorbent traps containing HgCl2 at the lower bound mass loading level and the front section of three traps containing HgCl2 at the upper bound mass loading level. HgCl2 can be spiked as a gas, or as a liquid solution containing HgCl2. However the liquid volume spiked must be <100 μL. Prepare and analyze each spiked trap, using the techniques that will be used to prepare and analyze the field samples. The average recovery for three traps at each spike concentration must be between 90 and 110 percent. Again, if multiple types of sorbent media are to be analyzed, a separate analytical bias test is required for each sorbent material.
8.2.4 Determination of Target Sample Volume. The target sample volume is an estimate of the sample volume needed to ensure that valid emissions data are collected (i.e., that sample mass Hg loadings fall within the analytical calibration curve and are within the upper and lower bounds set by the analytical bias tests). The target sample volume and minimum sample mass can also be determined by performing a diagnostic test run prior to initiation of formal testing.
Example: If the minimum sample mass is 50 ng and the concentration of mercury in the stack gas is estimated to be 2 μg/m3 (ng/L) then the following calculation would be used to determine the target sample volume:
Target Sample Volume = (50 ng)/(2 ng/L) = 25 L
Note: For the purposes of relative accuracy testing of Hg monitoring systems under part 75 of this chapter and Performance Specification 12A in appendix B to this part, when the stack gas Hg concentration is expected to be very low (<0.5 μg/dscm) you may estimate the Hg concentration at 0.5 μg/dscm.
8.2.5 Determination of Sample Run Time. Sample run time will be a function of minimum sample mass (see Section 8.2.2), target sample volume and nominal equipment sample flow rate. The minimum sample run time for conducting relative accuracy test audits of Hg monitoring systems is 30 minutes and for emissions testing to characterize an emission source is 1 hour. The target sample run time can be calculated using the following example.
Example: If the target sample volume has been determined to be 25 L, then the following formula would be used to determine the sampling time necessary to acquire 25 L of gas when sampling at a rate of 0.4 L/min.
Sampling time (min) = 25 L / 0.4 L/min = 63 minutes
8.2.6 Field Recovery Test. The field recovery test provides a test program-specific verification of the performance of the combined sampling and analytical approach. Three sets of paired samples, one of each pair which is spiked with a known level of Hg, are collected and analyzed and the average recovery of the spiked samples is used to verify performance of the measurement system under field conditions during that test program. The conduct of this test requires an estimate or confirmation of the stack Hg concentrations at the time of testing.
8.2.6.1 Calculation of Pre-sampling Spiking Level. Determine the sorbent trap spiking level for the field recovery test using estimates of the stack Hg concentration, the target sample flow rate, and the planned sample duration. First, determine the Hg mass expected to be collected in section 1 of the sorbent trap. The pre-sampling spike must be within 50 to 150 percent of this expected mass.
Example calculation: For an expected stack Hg concentration of 5 ug/m3 (ng/L) a target sample rate of 0.40 liters/min, and a sample duration of 1 hour:
(0.40 L/min)*(60 min)*(5ng/L) = 120 ng
A Hg spike of 60 to 180 ng (50-150% of 120 ng) would be appropriate.
8.2.6.2 Procedures. Set up two identical sampling trains. One of the sampling trains shall be designated the spiked train and the other the unspiked train. Spike Hg0 onto the front section of the sorbent trap in the spiked train before sampling. The mass of Hg spiked shall be 50 to 150 percent of the mass expected to be collected with the unspiked train. Sample the stack gas with the two trains simultaneously using the same procedures as for the field samples (see Section 8.3). The total sample volume must be within ±20 percent of the target sample volume for the field sample test runs. Analyze the sorbent traps from the two trains utilizing the same analytical procedures and instrumentation as for the field samples (see Section 11.0). Determine the fraction of spiked Hg recovered (R) using the equations in Section 12.7. Repeat this procedure for a total of three runs. Report the individual R values in the test report; the average of the three R values must be between 85 and 115 percent.
Note to section 8.2.6.2: It is acceptable to perform the field recovery test concurrent with actual test runs (e.g., through the use of a quad probe). It is also acceptable to use the field recovery test runs as test runs for emissions testing or for the RATA of a Hg monitoring system under part 75 of this chapter and Performance Specification 12A in appendix B to this part, if certain conditions are met. To determine whether a particular field recovery test run may be used as a RATA run, subtract the mass of the Hg0 spike from the total Hg mass collected in sections 1 and 2 of the spiked trap. The difference represents the mass of Hg in the stack gas sample. Divide this mass by the sample volume to obtain the Hg concentration in the effluent gas stream, as measured with the spiked trap. Compare this concentration to the corresponding Hg concentration measured with the unspiked trap. If the paired trains meet the relative deviation and other applicable data validation criteria in Table 9-1, then the average of the two Hg concentrations may be used as an emissions test run value or as the reference method value for a RATA run.
8.3 Sampling. This section describes the procedures and criteria for collecting the field samples for analysis. As noted in Section 8.2.6, the field recovery test samples are also collected using these procedures.
8.3.1 Pre-test leak check. Perform a leak check of the sampling system with the sorbent traps in place. For each of the paired sampling trains, draw a vacuum in the train, and adjust the vacuum to ~15″ Hg; and, using the gas flow meter, determine leak rate. The leak rate for an individual train must not exceed 4 percent of the target sampling rate. Once the leak check passes this criterion, carefully release the vacuum in the sample train, then seal the sorbent trap inlet until the probe is ready for insertion into the stack or duct.
8.3.2 Determination of Flue Gas Characteristics. Determine or measure the flue gas measurement environment characteristics (gas temperature, static pressure, gas velocity, stack moisture, etc.) in order to determine ancillary requirements such as probe heating requirements (if any), initial sampling rate, moisture management, etc.
8.3.3 Sample Collection
8.3.3.1 Remove the plug from the end of each sorbent trap and store each plug in a clean sorbent trap storage container. Remove the stack or duct port cap and insert the probe(s). Secure the probe(s) and ensure that no leakage occurs between the duct and environment.
8.3.3.2 Record initial data including the sorbent trap ID, date, and the run start time.
8.3.3.3 Record the initial gas flow meter reading, stack temperature, meter temperatures (if needed), and any other appropriate information, before beginning sampling. Begin sampling and target a sampling flow rate similar to that for the field recovery test. Then, at regular intervals (≤5 minutes) during the sampling period, record the date and time, the sample flow rate, the gas meter reading, the stack temperature, the flow meter temperatures (if using a dry gas meter), temperatures of heated equipment such as the vacuum lines and the probes (if heated), and the sampling system vacuum readings. Adjust the sampling flow rate as necessary to maintain the initial sample flow Start Printed Page 51522rate. Ensure that the total volume sampled for each run is within 20 percent of the total volume sampled for the field recovery test.
8.3.3.4 Data Recording. Obtain and record any essential operating data for the facility during the test period, e.g., the barometric pressure must be obtained for correcting sample volume to standard conditions when using a dry gas meter. At the end of the data collection period, record the final gas flow meter reading and the final values of all other essential parameters.
8.3.3.5 Post-Test Leak Check. When sampling is completed, turn off the sample pump, remove the probe(s) with sorbent traps from the port, and carefully seal the end of each sorbent trap. Perform another leak check of each sampling train with the sorbent trap in place, at the maximum vacuum reached during the sampling period. Record the leakage rates and vacuums. The leakage rate for each train must not exceed 4 percent of the average sampling rate for the data collection period. Following each leak check, carefully release the vacuum in the sample train.
8.3.3.6 Sample Recovery. Recover each sampled sorbent trap by removing it from the probe and sealing both ends. Wipe any deposited material from the outside of the sorbent trap. Place the sorbent trap into an appropriate sample storage container and store/preserve in appropriate manner (see Section 8.3.3.8).
8.3.3.7 Stack Gas Moisture Determination. If the moisture basis of the measurements made with this method (dry) is different from the moisture basis of either: (1) the applicable emission limit; or (2) a Hg CEMS being evaluated for relative accuracy, you must determine the moisture content of the flue gas and correct for moisture using Method 4 in appendix A-3 to this part. If correction of the measured Hg concentrations for moisture is required, at least one Method 4 moisture determination shall be made during each test run.
8.3.3.8 Sample Handling, Preservation, Storage, and Transport. While the performance criteria of this approach provide for verification of appropriate sample handling, it is still important that the user consider, determine, and plan for suitable sample preservation, storage, transport, and holding times for these measurements. Therefore, procedures in ASTM WK223 “Guide for Packaging and Shipping Environmental Samples for Laboratory Analysis” shall be followed for all samples, where appropriate. To avoid Hg contamination of the samples, special attention should be paid to cleanliness during transport, field handling, sampling, recovery, and laboratory analysis, as well as during preparation of the sorbent cartridges. Collection and analysis of blank samples (e.g., reagent, sorbent, field, etc.,) is useful in verifying the absence or source of contaminant Hg.
8.3.3.9 Sample Custody. Proper procedures and documentation for sample chain of custody are critical to ensuring data integrity. The chain of custody procedures in ASTM D4840-99 “Standard Guide for Sampling Chain-of-Custody Procedures” shall be followed for all samples (including field samples and blanks).
9.0 Quality Assurance and Quality Control
Table 9-1 summarizes the QA/QC performance criteria that are used to validate the Hg emissions data from Method 30B sorbent trap measurement systems.
Table 9-1.—Quality Assurance/Quality Control Criteria for Method 30B
QA/QC test or specification Acceptance criteria Frequency Consequences if not met Gas flow meter calibration (At 3 settings or points) Calibration factor (Yi) at each flow rate must be within ± 2% of the average value (Y) Prior to initial use and when post-test check is not within ± 5% of Y Recalibrate at 3 points until the acceptance criteria are met. Gas flow meter post-test calibration check (Single-point) Calibration factor (Yi) must be within ± 5% of the Y value from the most recent 3-point calibration After each field test. For mass flow meters, must be done on-site, using stack gas Recalibrate gas flow meter at 3 points to determine a new value of Y. For mass flow meters, must be done on-site, using stack gas. Apply the new Y value to the field test data. Temperature sensor calibration Absolute temperature measures by sensor within ± 1.5% of a reference sensor Prior to initial use and before each test thereafter Recalibrate; sensor may not be used until specification is met. Barometer calibration Absolute pressure measured by instrument within ± 10 mm Hg of reading with a mercury barometer Prior to initial use and before each test thereafter Recalibrate; instrument may not be used until specification is met. Pre-test leak check ≤ 4% of target sampling rate Prior to sampling Sampling shall not commence until the leak check is passed. Post-test leak check ≤ 4% of average sampling rate After sampling Sample invalidated.* Analytical matrix interference test (wet chemical analysis, only) Establish minimum dilution (if any) needed to eliminate sorbent matrix interferences Prior to analyzing any field samples; repeat for each type of sorbent used Field sample results not validated. Analytical bias test Average recovery between 90% and 110% for Hg0 and HgCl2 at each of the 2 spike concentration levels Prior to analyzing field samples and prior to use of new sorbent media Field samples shall not be analyzed until the percent recovery criteria has been met. Multipoint analyzer calibration Each analyzer reading withini ± 10% of true value and r2 ≥ 0.99 On the day of analysis, before analyzing any samples Recalibrate until successful. Analysis of independent calibration standard Within ± 10% of true value Following daily calibration, prior to analyzing field samples Recalibrate and repeat independent standard analysis until successful. Analysis of continuing calibration verification standard (CCVS) Within ± 10% of true value Following daily calibration, after analyzing ≤10 field samples, and at end of each set of analyses Recalibrate and repeat independent standard analysis, reanalyze samples until successful, if possible; for destructive techniques, samples invalidated. Test run total sample volume Within ± 20% of total volume sampled during field recovery test Each individual sample Sample invalidated. Sorbent trap section 2 breakthrough <10% of section 1 Hg mass for Hg concentrations > 1 μg/dscm; Every sample Sample invalidated.* Start Printed Page 51523 ≤ 20% of section 1 Hg mass for Hg concentrations ≤ 1 μg/dscm Paired sorbent trap agreement ≤ 10% Relative Deviation (RD) mass for Hg concentrations > 1 μg/dscm; Every run Run invalidated.* ≤ 20% RD or ≤ 0.2 μg/dscm absolute difference for Hg concentrations ≤ 1 μg/dscm Sample analysis Within valid calibration range (within calibration curve) All Section 1 samples where stack Hg concentation is ≥ 0.5 μg/dscm Reanalyze at more concentrated level if possible, samples invalidated if not within calibrated range. Sample analysis Within bounds of Hg0 and HgCl2 Analytical Bias Test All Section 1 samples where stack Hg concentration is ≥ 0.5 μg/dscm Expand bounds of Hg0 and HgCl2 Analytical Bias Test; if not successful, samples invalidated. Field recovery test Average recovery between 85% and 115% for Hg0 Once per field test Field sample runs not validated without successful field recovery test. * And data from the pair of sorbent traps are also invalidated. 10.0 Calibration and Standardization
10.1 Only NIST-certified and NIST-traceable calibration standards (i.e., calibration gases, solutions, etc.) shall be used for the spiking and analytical procedures in this method.
10.2 Gas Flow Meter Calibration.
10.2.1 Preliminaries. The manufacturer or equipment supplier of the gas flow meter should perform all necessary set-up, testing, programming, etc., and should provide the end user with any necessary instructions, to ensure that the meter will give an accurate readout of dry gas volume in standard cubic meters for this method.
10.2.2 Initial Calibration. Prior to its initial use, a calibration of the gas flow meter shall be performed. The initial calibration may be done by the manufacturer, by the equipment supplier, or by the end user. If the flow meter is volumetric in nature (e.g., a dry gas meter), the manufacturer or end user may perform a direct volumetric calibration using any gas. For a mass flow meter, the manufacturer, equipment supplier, or end user may calibrate the meter using either: (1) A bottled gas mixture containing 12 ±0.5% CO2, 7 ±0.5% O2, and balance N2 (when this method is applied to coal-fired boilers); (2) a bottled gas mixture containing CO2, O2, and N2 in proportions representative of the expected stack gas composition; or (3) the actual stack gas.
10.2.2.1 Initial Calibration Procedures. Determine an average calibration factor (Y) for the gas flow meter by calibrating it at three sample flow rate settings covering the range of sample flow rates at which the sampling system will be operated. You may either follow the procedures in section 10.3.1 of Method 5 in appendix A-3 to this part or in section 16 of Method 5 in appendix A-3 to this part. If a dry gas meter is being calibrated, use at least five revolutions of the meter at each flow rate.
10.2.2.2 Alternative Initial Calibration Procedures. Alternatively, you may perform the initial calibration of the gas flow meter using a reference gas flow meter (RGFM). The RGFM may be: (1) A wet test meter calibrated according to section 10.3.1 of Method 5 in appendix A-3 to this part; (2) a gas flow metering device calibrated at multiple flow rates using the procedures in section 16 of Method 5 in appendix A-3 to this part; or (3) a NIST-traceable calibration device capable of measuring volumetric flow to an accuracy of 1 percent. To calibrate the gas flow meter using the RGFM, proceed as follows: While the Method 30B sampling system is sampling the actual stack gas or a compressed gas mixture that simulates the stack gas composition (as applicable), connect the RGFM to the discharge of the system. Care should be taken to minimize the dead volume between the gas flow meter being tested and the RGFM. Concurrently measure dry stack gas volume with the RGFM and the flow meter being calibrated for at least 10 minutes at each of three flow rates covering the typical range of operation of the sampling system. For each set of concurrent measurements, record the total sample volume, in units of dry standard cubic meters (dscm), measured by the RGFM and the gas flow meter being tested.
10.2.2.3 Initial Calibration Factor. Calculate an individual calibration factor Yi at each tested flow rate from section 10.2.2.1 or 10.2.2.2 of this method (as applicable) by taking the ratio of the reference sample volume to the sample volume recorded by the gas flow meter. Average the three Yi values, to determine Y, the calibration factor for the flow meter. Each of the three individual values of Yi must be within ±0.02 of Y. Except as otherwise provided in sections 10.2.2.4 and 10.2.2.5 of this method, use the average Y value from the initial 3-point calibration to adjust subsequent gas volume measurements made with the gas flow meter.
10.2.2.4 Pretest On-Site Calibration Check (Optional). For a mass flow meter, if the most recent 3-point calibration of the flow meter was performed using a compressed gas mixture, you may want to conduct the following on-site calibration check prior to testing, to ensure that the flow meter will accurately measure the volume of the stack gas: While sampling stack gas, check the calibration of the flow meter at one intermediate flow rate setting representative of normal operation of the sampling system. If the pretest calibration check shows that the value of Yi, the calibration factor at the tested flow rate, differs from the current value of Y by more than 5 percent, perform a full 3-point recalibration of the meter using stack gas to determine a new value of Y, and (except as otherwise provided in section 10.2.2.5 of this method) apply the new Y value to the data recorded during the field test.
10.2.2.5 Post-Test Calibration Check. Check the calibration of the gas flow meter following each field test at one intermediate flow rate setting, either at, or in close proximity to, the average sample flow rate during the field test. For dry gas meters, ensure at least three revolutions of the meter during the calibration check. For mass flow meters, this check must be performed before leaving the test site, while sampling stack gas. If a one-point calibration check shows that the value of Yi at the tested flow rate differs by more than 5 percent from the current value of Y, repeat the full 3-point calibration procedure to determine a new value of Y, and apply the new Y value to the gas volume measurements made with the gas flow meter during the field test that was just completed. For mass flow meters, perform the 3-point recalibration while sampling stack gas.
10.3 Thermocouples and Other Temperature Sensors. Use the procedures and criteria in Section 10.3 of Method 2 in Appendix A-1 to this part to calibrate in-stack temperature sensors and thermocouples. Dial thermometers shall be calibrated against mercury-in-glass thermometers. Calibrations must be performed prior to initial use and before each field test thereafter. At each calibration point, the absolute temperature measured by the temperature sensor must agree to within ±1.5 percent of the temperature measured with the reference sensor, otherwise the sensor may not continue to be used. Start Printed Page 51524
10.4 Barometer. Calibrate against a mercury barometer as per Section 10.6 of Method 5 in appendix A-3 to this part. Calibration must be performed prior to initial use and before each test program, and the absolute pressure measured by the barometer must agree to within +10 mm Hg of the pressure measured by the mercury barometer, otherwise the barometer may not continue to be used.
10.5 Other Sensors and Gauges. Calibrate all other sensors and gauges according to the procedures specified by the instrument manufacturer(s).
10.6 Analytical System Calibration. See Section 11.1 of this method.
11.0 Analytical Procedures
The analysis of Hg in the field and quality control samples may be conducted using any instrument or technology capable of quantifying total Hg from the sorbent media and meeting the performance criteria in this method. Because multiple analytical approaches, equipment and techniques are appropriate for the analysis of sorbent traps, it is not possible to provide detailed, technique-specific analytical procedures. As they become available, detailed procedures for a variety of candidate analytical approaches will be posted at http://www.epa.gov/ttn/emc.
11.1 Analytical System Calibration. Perform a multipoint calibration of the analyzer at three or more upscale points over the desired quantitative range (multiple calibration ranges shall be calibrated, if necessary). The field samples analyzed must fall within a calibrated, quantitative range and meet the performance criteria specified below. For samples suitable for aliquotting, a series of dilutions may be needed to ensure that the samples fall within a calibrated range. However, for sorbent media samples consumed during analysis (e.g., when using thermal desorption techniques), extra care must be taken to ensure that the analytical system is appropriately calibrated prior to sample analysis. The calibration curve range(s) should be determined such that the levels of Hg mass expected to be collected and measured will fall within the calibrated range. The calibration curve may be generated by directly introducing standard solutions into the analyzer or by spiking the standards onto the sorbent media and then introducing into the analyzer after preparing the sorbent/standard according to the particular analytical technique. For each calibration curve, the value of the square of the linear correlation coefficient, i.e., r2, must be ≥0.99, and the analyzer response must be within ±10 percent of the reference value at each upscale calibration point. Calibrations must be performed on the day of the analysis, before analyzing any of the samples. Following calibration, an independent standard shall be analyzed. The measured value of the independently prepared standard must be within ±10 percent of the expected value.
11.2 Sample Preparation. Carefully separate the sections of each sorbent trap. Combine for analysis all materials associated with each section; any supporting substrate that the sample gas passes through prior to entering a media section (e.g., glass wool separators, acid gas traps, etc.) must be analyzed with that segment.
11.3 Field Sample Analyses. Analyze the sorbent trap samples following the same procedures that were used for conducting the Hg0 and HgCl2 analytical bias tests. The individual sections of the sorbent trap and their respective components must be analyzed separately (i.e., section 1 and its components, then section 2 and its components). All sorbent trap section 1 sample analyses must be within the calibrated range of the analytical system. For wet analyses, the sample can simply be diluted to fall within the calibrated range. However, for the destructive thermal analyses, samples that are not within the calibrated range cannot be re-analyzed. As a result, the sample cannot be validated, and another sample must be collected. It is strongly suggested that the analytical system be calibrated over multiple ranges so that thermally analyzed samples do fall within the calibrated range. The total mass of Hg measured in each sorbent trap section 1 must also fall within the lower and upper mass limits established during the initial Hg0 and HgCl2 analytical bias test. If a sample is analyzed and found to fall outside of these limits, it is acceptable for an additional Hg0 and HgCl2 analytical bias test to be performed that now includes this level. However, some samples (e.g., the mass collected in trap section 2 or the mass collected in trap section 1 when the stack gas concentration is <0.5 μg/m3), may have Hg levels so low that it may not be possible to quantify them in the analytical system's calibrated range. Because a reliable estimate of these low-level Hg measurements is necessary to fully validate the emissions data, the MDL (see section 8.2.2.1 of this method) is used to establish the minimum amount that can be detected and reported. If the measured mass or concentration is below the lowest point in the calibration curve and above the MDL, the analyst must do the following: estimate the mass or concentration of the sample based on the analytical instrument response relative to an additional calibration standard at a concentration or mass between the MDL and the lowest point in the calibration curve. This is accomplished by establishing a response factor (e.g., area counts per Hg mass or concentration) and estimating the amount of Hg present in the sample based on the analytical response and this response factor.
Example: The analysis of a particular sample results in a measured mass above the MDL, but below the lowest point in the calibration curve which is 10 ng. An MDL of 1.3 ng Hg has been established by the MDL study. A calibration standard containing 5 ng of Hg is analyzed and gives an analytical response of 6,170 area counts, which equates to a response factor of 1,234 area counts/ng Hg. The analytical response for the sample is 4,840 area counts. Dividing the analytical response for the sample (4,840 area counts) by the response factor gives 3.9 ng Hg, which is the estimated mass of Hg in the sample.
11.4 Analysis of Continuing Calibration Verification Standard (CCVS). After no more than 10 samples and at the end of each set of analyses, a continuing calibration verification standard must be analyzed. The measured value of the continuing calibration standard must be within ±10 percent of the expected value.
11.5 Blanks. The analysis of blanks is optional. The analysis of blanks is useful to verify the absence of, or an acceptable level of, Hg contamination. Blank levels should be considered when quantifying low Hg levels and their potential contribution to meeting the sorbent trap section 2 breakthrough requirements; however, correcting sorbent trap results for blank levels is prohibited.
12.0 Calculations and Data Analysis
You must follow the procedures for calculation and data analysis listed in this section.
12.1 Nomenclature. The terms used in the equations are defined as follows:
B = Breakthrough (%).
Bws = Moisture content of sample gas as measured by Method 4, percent/100.
Ca = Concentration of Hg for the sample collection period, for sorbent trap “a” (μg/dscm).
Cb = Concentration of Hg for the sample collection period, for sorbent trap “b” (μg/dscm).
Cd = Hg concentration, dry basis (μg/dscm).
Crec = Concentration of spiked compound measured (μg/m3).
Cw = Hg concentration, wet basis (μg/m3).
m1 = Mass of Hg measured on sorbent trap section 1 (μg).
m2 = Mass of Hg measured on sorbent trap section 2 (μg).
mrecovered = Mass of spiked Hg recovered in Analytical Bias or Field Recovery Test (μg).
ms = Total mass of Hg measured on spiked trap in Field Recovery Test (μg).
mspiked = Mass of Hg spiked in Analytical Bias or Field Recovery Test (μg).
mu = Total mass of Hg measured on unspiked trap in Field Recovery Test (μg).
R = Percentage of spiked mass recovered (%).
RD = Relative deviation between the Hg concentrations from traps “a” and “b” (%).
vs = Volume of gas sampled, spiked trap in Field Recovery Test (dscm).
Vt = Total volume of dry gas metered during the collection period (dscm); for the purposes of this method, standard temperature and pressure are defined as 20 °C and 760 mm Hg, respectively.
vu = Volume of gas sampled, unspiked trap in Field Recovery Test (dscm).
12.2 Calculation of Spike Recovery (Analytical Bias Test). Calculate the percent recovery of Hg0 and HgCl2 using Equation 30B-1.

12.3 Calculation of Breakthrough. Use Equation 30B-2 to calculate the percent breakthrough to the second section of the sorbent trap.
Start Printed Page 51525
12.4 Calculation of Hg Concentration. Calculate the Hg concentration measured with sorbent trap “a”, using Equation 30B-3.

For sorbent trap “b”, replace “Ca ” with “Cb ” in Equation 30B-3. Report the average concentration, i.e., 1/2 (Ca + Cb).
12.5 Moisture Correction. Use Equation 30B-4 if your measurements need to be corrected to a wet basis.

12.6 Calculation of Paired Trap Agreement. Calculate the relative deviation (RD) between the Hg concentrations measured with the paired sorbent traps using Equation 30B-5.

12.7 Calculation of Measured Spike Hg Concentration (Field Recovery Test). Calculate the measured spike concentration using Equation 30B-6.

Then calculate the spiked Hg recovery, R, using Equation 30B-7.

13.0 Method Performance
How do I validate my data? Measurement data are validated using initial, one-time laboratory tests coupled with test program-specific tests and procedures. The analytical matrix interference test and the Hg0 and HgCl2 analytical bias test described in Section 8.2 are used to verify the appropriateness of the selected analytical approach(es) as well as define the valid working ranges for sample analysis. The field recovery test serves to verify the performance of the combined sampling and analysis as applied for each test program. Field test samples are validated by meeting the above requirements as well as meeting specific sampling requirements (i.e., leak checks, paired train agreement, total sample volume agreement with field recovery test samples) and analytical requirements (i.e., valid calibration curve, continuing calibration performance, sample results within calibration curve and bounds of Hg0 and HgCl2 analytical bias test). Complete data validation requirements are summarized in Table 9-1.
14.0 Pollution Prevention [Reserved]
15.0 Waste Management [Reserved]
16.0 References
1. EPA Traceability Protocol for Qualification and Certification of Elemental Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
2. EPA Traceability Protocol for Qualification and Certification of Oxidized Mercury Gas Generators, expected publication date December 2008, see www.epa.gov/ttn/emc.
3. EPA Traceability Protocol for Assay and Certification of Gaseous Calibration Standards, expected revision publication date December 2008, see www.epa.gov/ttn/emc.
17.0 Figures and Tables
Start Printed Page 51526
Start Printed Page 51527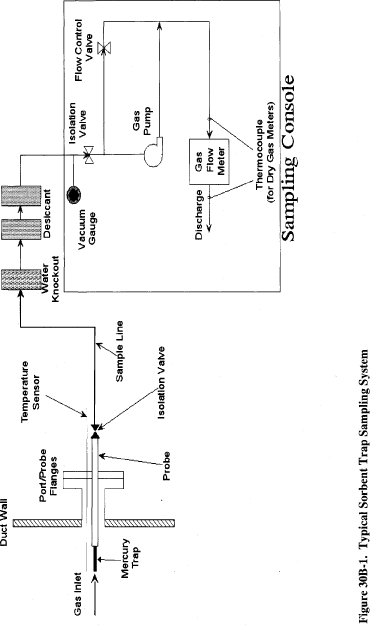
Appendix B [Amended]
Start Amendment Part3. Amend Performance Specification 12A in Appendix B to part 60 by revising sections 8.6.2, 8.6.4, 8.6.5, and 8.6.6.1 to read as follows:
End Amendment PartPerformance Specification 12A—Specifications and Test Procedures for Total Vapor Phase Mercury Continuous Emission Monitoring Systems in Stationary Sources
* * * * *8.6.2 RM. Unless otherwise specified in an applicable subpart of the regulations, use Method 29, Method 30A, or Method 30B in appendix A to this part or American Society of Testing and Materials (ASTM) Method D6784-02 (incorporated by reference, see § 60.17) as the RM for Hg concentration. Do not include the filterable portion of the sample when making comparisons to the CEMS results. When Method 29, Method 30B, or ASTM D6784-02 is used, conduct the RM test runs with paired or duplicate sampling systems. When Method 30A is used, paired sampling systems are not required. If the RM and CEMS measure on a different moisture basis, data derived with Method 4 in appendix A to this part shall also be obtained during the RA test.
* * * * *8.6.4 Number and Length of RM and Tests. Conduct a minimum of nine RM test runs. When Method 29, Method 30B, or ASTM D6784-02 is used, only test runs for which the paired RM trains meet the relative deviation criteria (RD) of this PS shall be used in the RA calculations. In addition, for Method 29 and ASTM D6784-02, use a minimum sample time of 2 hours and for Method 30A use a minimum sample time of 30 minutes.
Note:
More than nine sets of RM tests may be performed. If this option is chosen, paired RM test results may be excluded so long as the total number of paired RM test results used to determine the CEMS RA is greater than or equal to nine. However, all data must be reported including the excluded data.
8.6.5 Correlation of RM and CEMS Data. Correlate the CEMS and the RM test data as to the time and duration by first determining from the CEMS final output (the one used for reporting) the integrated average pollutant concentration for each RM test period. Consider system response time, if important, and confirm that the results are on a consistent moisture basis with the RM test. Then, compare each integrated CEMS value against the corresponding RM value. When Method 29, Method 30A, Method 30B, or ASTM D6784-02 is used, compare each CEMS value against the corresponding average of the paired RM values.
8.6.6 * * *
8.6.6.1 When Method 29, Method 30B, or ASTM D6784-02 is used, outliers are identified through the determination of relative deviation (RD) of the paired RM tests. Data that do not meet the criteria should be flagged as a data quality problem. The primary reason for performing paired RM sampling is to ensure the quality of the RM data. The percent RD of paired data is the parameter used to quantify data quality. Determine RD for two paired data points as follows:

where Ca and Cb are concentration values determined from each of the two samples, respectively.
* * * * *Start PartPART 72—PERMITS REGULATION
End Part Start Amendment Part4. The authority citation for part 72 continues to read as follows:
End Amendment Part Start Amendment Part5. Revise the definition of “sorbent trap monitoring system” in § 72.2 as follows:
End Amendment PartStart PartDefinitions.* * * * *Sorbent trap monitoring system means the equipment required by part 75 of this chapter for the continuous monitoring of Hg emissions, using paired sorbent traps containing iodated charcoal (IC) or other suitable reagents. This excepted monitoring system consists of a probe, the paired sorbent traps, an umbilical line, moisture removal components, an air tight sample pump, a gas flow meter, and an automated data acquisition and handling system. The monitoring system samples the stack gas at a rate proportional to the stack gas volumetric flowrate. The sampling is a batch process. Using the sample volume measured by the gas flow meter and the results of the analyses of the sorbent traps, the average mercury concentration in the stack gas for the sampling period is determined, in units of micrograms per dry standard cubic meter (μg/dscm). Mercury mass emissions for each hour in the sampling period are calculated using the average Hg concentration for that period, in conjunction with contemporaneous hourly measurements of the stack gas flow rate, corrected for the stack moisture content.
* * * * *PART 75—CONTINUOUS EMISSION MONITORING
End Part Start Amendment Part6. The authority citation for part 75 continues to read as follows:
End Amendment Part Start Amendment Part7. Amend § 75.15 as follows:
End Amendment Part Start Amendment Parta. Revise paragraph (f);
End Amendment Part Start Amendment Partb. Revise paragraph (i); and
End Amendment Part Start Amendment Partc. Add new paragraph (k).
End Amendment PartThe revisions and additions read as follows:
Start Amendment PartSpecial provisions for measuring Hg mass emissions using the excepted sorbent trap monitoring methodology.* * * * *(f) At the beginning and end of each sample collection period, and at least once in each unit operating hour during the collection period, the gas flow meter reading shall be recorded.
* * * * *(i) All unit operating hours for which valid Hg concentration data are obtained with the primary sorbent trap monitoring system (as verified using the quality assurance procedures in appendix K to this part) shall be reported in the electronic quarterly report under § 75.84(f). For hours in which data from the primary monitoring system are invalid, the owner or operator may, in accordance with § 75.20(d), report valid Hg concentration data from: A certified redundant backup CEMS or sorbent trap monitoring system; a certified non-redundant backup CEMS or sorbent trap monitoring system; or an applicable reference method under § 75.22. If no quality-assured Hg concentration are available for a particular hour, the owner or operator shall report the appropriate substitute data value in accordance with § 75.39.
* * * * *(k) During each RATA of a sorbent trap monitoring system, the type of sorbent material used by the traps shall be the same as for daily operation of the monitoring system. A new pair of traps shall be used for each RATA run. However, the size of the traps used for the RATA may be smaller than the traps used for daily operation of the system.
* * * * *8. Amend § 75.20 by adding new paragraph (d)(2)(ix) to read as follows:
End Amendment PartStart Amendment PartInitial certification and recertification procedures.* * * * *(d)* * *
(2)* * *
(ix) For non-redundant backup Hg CEMS and sorbent trap monitoring systems, and for like-kind replacement Hg analyzers, the following provisions apply in addition to, or, in some cases, in lieu of, the general requirements in paragraphs (d)(2)(i) through (d)(2)(viii) of this section:
(A) When a certified sorbent trap monitoring system is brought into service as a regular non-redundant backup monitoring system, the system shall be operated according to the procedures in § 75.15 and appendix K of this part;
(B) When a regular non-redundant backup Hg CEMS or a like-kind Start Printed Page 51528replacement Hg analyzer is brought into service, a linearity check with elemental Hg standards, as described in paragraph (c)(1)(ii) of this section and section 6.2 of appendix A of this part, and a single-point system integrity check, as described in section 2.6 of appendix B of this part, shall be performed. Alternatively, a 3-level system integrity check, as described in paragraph (c)(1)(vi) of this section and paragraph (g) of section 6.2 in appendix A of this part, may be performed in lieu of these two tests.
(C) The weekly single-point system integrity checks described in section 2.6 of appendix B of this part are required as long as a non-redundant backup Hg CEMS or like-kind replacement Hg analyzer remains in service, unless the daily calibrations of the Hg analyzer are done using a NIST-traceable source of oxidized Hg.
* * * * *9. Amend § 75.57 by revising paragraph (j)(7) to read as follows:
End Amendment PartStart Amendment PartGeneral recordkeeping provisions.* * * * *(j) * * *
(7) Record the gas flow meter reading (in dscm, rounded to the nearest hundreth) at the beginning and end of the collection period and at least once in each unit operating hour during the collection period.
* * * * *10. Amend § 75.81 by revising paragraph (a)(1) to read as follows:
End Amendment PartStart Amendment PartMonitoring of Hg mass emissions and heat input at the unit level.* * * * *(a) * * *
(1) A Hg concentration monitoring system (as defined in § 72.2 of this chapter) or a sorbent trap monitoring system (as defined in § 72.2 of this chapter), to measure the mass concentration of total vapor phase Hg in the flue gas, including the elemental and oxidized forms of Hg, in micrograms per standard cubic meter (μg/scm); and
* * * * *11. Amend § 75.84 by revising paragraph (f)(1)(ii)(J) to read as follows:
End Amendment PartStart Amendment PartRecordkeeping and Reporting.* * * * *(f) * * *
(1) * * *
(ii) * * *
(J) For units using sorbent trap monitoring systems, the hourly gas flow meter readings taken between the initial and final meter readings for the data collection period; and
* * * * *Appendix A to Part 75—[Amended]
12. Amend Appendix A to part 75 by removing the twentieth sentence in paragraph (a) of section 6.5.7 which currently reads “For the RATA of a sorbent trap monitoring system, use the same size trap that is used for daily operation of the monitoring system.” and adding in its place “For the RATA of a sorbent trap monitoring system, the type of sorbent material used by the traps shall be the same as for daily operation of the monitoring system; however, the size of the traps used for the RATA may be smaller than the traps used for daily operation of the system.”.
End Amendment Part Start Amendment Part13. Amend Appendix B to part 75 by revising section 1.5.2 to read as follows:
End Amendment PartAppendix B to Part 75—Quality Assurance and Quality Control Procedures
* * * * *1.5.2 Monitoring System Integrity and Data Quality
Explain the procedures used to perform the leak checks when sorbent traps are placed in service and removed from service. Also explain the other QA procedures used to ensure system integrity and data quality, including, but not limited to, gas flow meter calibrations, verification of moisture removal, and ensuring air-tight pump operation. In addition, the QA plan must include the data acceptance and quality control criteria in section 8 of appendix K to this part. All reference meters used to calibrate the gas flow meters (e.g., wet test meters) shall be periodically recalibrated. Annual, or more frequent, recalibration is recommended. If a NIST-traceable calibration device is used as a reference flow meter, the QA plan must include a protocol for ongoing maintenance and periodic recalibration to maintain the accuracy and NIST-traceability of the calibrator.
* * * * *Start Amendment Part14. Amend Appendix K to part 75 as follows:
End Amendment Part Start Amendment Parta. Amend section 5.1 by revising Figure K-1;
End Amendment Part Start Amendment Partb. Revise section 5.1.3;
End Amendment Part Start Amendment Partc. Revise section 5.1.5;
End Amendment Part Start Amendment Partd. Revise section 7.1.3;
End Amendment Part Start Amendment Parte. Revise section 7.2.3;
End Amendment Part Start Amendment Partf. Revise section 7.2.5;
End Amendment Part Start Amendment Partg. Amend section 8.0 by revising Table K-1;
End Amendment Part Start Amendment Parth. Revise section 9.2;
End Amendment Part Start Amendment Parti. Revise section 10.4;
End Amendment Part Start Amendment Partj. Remove and reserve section 11.5;
End Amendment Part Start Amendment Partk. Revise section 11.6; and
End Amendment Part Start Amendment Partl. Revise section 11.7.
End Amendment PartThe revisions and additions read as follows:
Start AppendixAppendix K to Part 75—Quality Assurance and Operating Procedures for Sorbent Trap Monitoring Systems
* * * * *5.1 * * *
Start Printed Page 51529
Start Printed Page 51530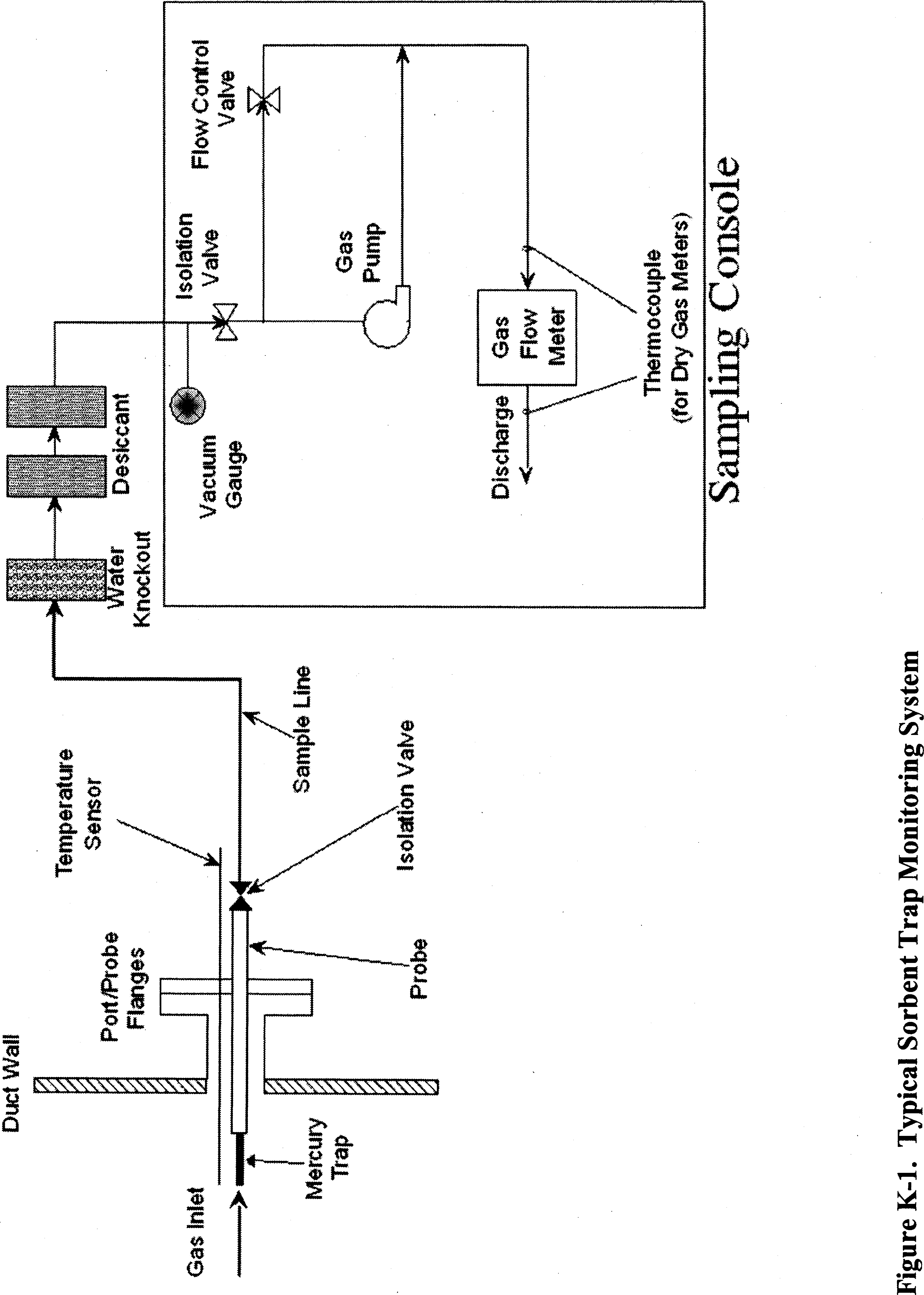 * * * * *
* * * * *5.1.3 Moisture Removal Device
A robust moisture removal device or system, suitable for continuous duty (such as a Peltier cooler), shall be used to remove water vapor from the gas stream prior to entering the gas flow meter.
* * * * *5.1.5 Gas Flow Meter
A gas flow meter (such as a dry gas meter, thermal mass flow meter, or other suitable measurement device) shall be used to determine the total sample volume on a dry basis, in units of standard cubic meters. The meter must be sufficiently accurate to measure the total sample volume to within 2 percent and must be calibrated at selected flow rates across the range of sample flow rates at which the sorbent trap monitoring system typically operates. The gas flow meter shall be equipped with any necessary auxiliary measurement devices (e.g., temperature sensors, pressure measurement devices) needed to correct the sample volume to standard conditions.
* * * * *7.1.3 Pre-test Leak Check
Perform a leak check with the sorbent traps in place. Draw a vacuum in each sample train. Adjust the vacuum in the sample train to ~15″ Hg. Using the gas flow meter, determine leak rate. The leakage rate must not exceed 4 percent of the target sampling rate. Once the leak check passes this criterion, carefully release the vacuum in the sample train then seal the sorbent trap inlet until the probe is ready for insertion into the stack or duct.
* * * * *7.2.3 Flow Rate Control
Set the initial sample flow rate at the target value from section 7.1.1 of this appendix. Record the initial gas flow meter reading, stack temperature (if needed to convert to standard conditions), meter temperatures (if needed), etc. Then, for every operating hour during the sampling period, record the date and time, the sample flow rate, the gas flow meter reading, the stack temperature (if needed), the flow meter temperatures (if needed), temperatures of heated equipment such as the vacuum lines and the probes (if heated), and the sampling system vacuum readings. Also, record the stack gas flow rate, as measured by the certified flow monitor, and the ratio of the stack gas flow rate to the sample flow rate. Adjust the sampling flow rate to maintain proportional sampling, i.e., keep the ratio of the stack gas flow rate to sample flow rate constant, to within ±25 percent of the reference ratio from the first hour of the data collection period (see section 11 of this appendix).
* * * * *7.2.5 Essential Operating Data
Obtain and record any essential operating data for the facility during the test period, e.g., the barometric pressure for correcting the sample volume measured by a dry gas meter to standard conditions. At the end of the data collection period, record the final gas flow meter reading and the final values of all other essential parameters.
* * * * *8.0 * * *
Table K-1.—Quality Assurance/Quality Control Criteria for Sorbent Trap Monitoring Systems
QA/QC test or specification Acceptance criteria Frequency Consequences if not met Pre-test leak check ≤4% of target sampling rate Prior to Sampling Sampling shall not commence until the leak check is passed. Post-test leak check ≤4% of average sampling rate After sampling Sample check invalidated.** Ratio of stack gas flow rate to sample flow rate Maintain within ±25% of initial ratio from first hour of data collection period Every hour throughout data collection period Case-by-case evaluation. Sorbent trap section 2 breakthrough ≤5% of Section 1 Hg mass Every sample Sample invalidated.** Paired sorbent trap agreement ≤10% Relative Deviation (RD) Every sample Sample invalidated.** Spike recovery study Average recovery between 85% and 115% for each of the 3 spike concentration levels Prior to analyzing field samples and prior to use of new sorbent media Field samples shall not be analyzed until the percent recovery criterion has been met. Multipoint analyzer calibration Each analyzer reading within ±10% of true value and r2 ≥0.99 On the day of analysis, before analyzing any samples Recalibrate until successful. Analysis of independent calibration standard Within ±10% of true value Following daily calibration, prior to analyzing field Recalibrate and repeat independent standard analysis samples until successful. Spike recovery from section 3 of sorbent trap 75-125% of spike amount Every sample Sample invalidated.** RATA RA ≤20.0% or Mean difference ≤1.0 μgm/dscm for low emitters For initial certification and annually thereafter Data from the system are invalidated until a RATA is passed. Gas flow meter calibration (At 3 settings initially, and 1 setting thereafter) Calibration factor (Y) within ±5% of average value from the initial (3-point) calibration Prior to initial use and at least quarterly thereafter Recalibrate the meter at three settings to determine a new value of Y. Temperature sensor calibration Absolute temperature measured by sensor within ±1.5% of a reference sensor Prior to initial use and at least quarterly thereafter Recalibrate. Sensor may not be used until specification is met. Barometer calibration Absolute pressure measured by instrument within ±10 mm Hg of reading with a mercury barometer Prior to initial use and at least quarterly thereafter Recalibrate. Instrument may not be used until specification is met. ** And data from the pair of sorbent traps are also invalidated. * * * * *9.2 Gas Flow Meter Calibration
9.2.1 Preliminaries. The manufacturer or supplier of the gas flow meter should perform all necessary set-up, testing, programming, etc., and should provide the end user with any necessary instructions, to ensure that the meter will give an accurate readout of dry gas volume in standard cubic meters for the particular field application.
9.2.2 Initial Calibration. Prior to its initial use, a calibration of the flow meter shall be performed. The initial calibration may be done by the manufacturer, by the equipment supplier, or by the end user. If the flow meter is volumetric in nature (e.g., a dry gas meter), the manufacturer, equipment supplier, or end user may perform a direct volumetric calibration using any gas. For a mass flow meter, the manufacturer, equipment supplier, or end user may calibrate the meter using a bottled gas mixture containing 12 ± 0.5% CO2, 7 ± 0.5% O2, and balance N2, or these Start Printed Page 51531same gases in proportions more representative of the expected stack gas composition. Mass flow meters may also be initially calibrated on-site, using actual stack gas.
9.2.2.1 Initial Calibration Procedures. Determine an average calibration factor (Y) for the gas flow meter, by calibrating it at three sample flow rate settings covering the range of sample flow rates at which the sorbent trap monitoring system typically operates. You may either follow the procedures in section 10.3.1 of Method 5 in appendix A-3 to part 60 of this chapter or the procedures in section 16 of Method 5 in appendix A-3 to part 60 of this chapter. If a dry gas meter is being calibrated, use at least five revolutions of the meter at each flow rate.
9.2.2.2 Alternative Initial Calibration Procedures. Alternatively, you may perform the initial calibration of the gas flow meter using a reference gas flow meter (RGFM). The RGFM may either be: (1) A wet test meter calibrated according to section 10.3.1 of Method 5 in appendix A-3 to part 60; (2) a gas flow metering device calibrated at multiple flow rates using the procedures in section 16 of Method 5 in appendix A-3 to part 60; or (3) a NIST-traceable calibration device capable of measuring volumetric flow to an accuracy of 1 percent. To calibrate the gas flow meter using the RGFM, proceed as follows: While the sorbent trap monitoring system is sampling the actual stack gas or a compressed gas mixture that simulates the stack gas composition (as applicable), connect the RGFM to the discharge of the system. Care should be taken to minimize the dead volume between the sample flow meter being tested and the RGFM. Concurrently measure dry gas volume with the RGFM and the flow meter being calibrated the for a minimum of 10 minutes at each of three flow rates covering the typical range of operation of the sorbent trap monitoring system. For each 10-minute (or longer) data collection period, record the total sample volume, in units of dry standard cubic meters (dscm), measured by the RGFM and the gas flow meter being tested.
9.2.2.3 Initial Calibration Factor. Calculate an individual calibration factor Yi at each tested flow rate from section 9.2.2.1 or 9.2.2.2 of this appendix (as applicable), by taking the ratio of the reference sample volume to the sample volume recorded by the gas flow meter. Average the three Yi values, to determine Y, the calibration factor for the flow meter. Each of the three individual values of Yi must be within ±0.02 of Y. Except as otherwise provided in sections 9.2.2.4 and 9.2.2.5 of this appendix, use the average Y value from the three level calibration to adjust all subsequent gas volume measurements made with the gas flow meter.
9.2.2.4 Initial On-Site Calibration Check. For a mass flow meter that was initially calibrated using a compressed gas mixture, an on-site calibration check shall be performed before using the flow meter to provide data for this part. While sampling stack gas, check the calibration of the flow meter at one intermediate flow rate typical of normal operation of the monitoring system. Follow the basic procedures in section 9.2.2.1 or 9.2.2.2 of this appendix. If the on-site calibration check shows that the value of Yi, the calibration factor at the tested flow rate, differs by more than 5 percent from the value of Y obtained in the initial calibration of the meter, repeat the full 3-level calibration of the meter using stack gas to determine a new value of Y, and apply the new Y value to all subsequent gas volume measurements made with the gas flow meter.
9.2.2.5 Ongoing Quality Assurance. Recalibrate the gas flow meter quarterly at one intermediate flow rate setting representative of normal operation of the monitoring system. Follow the basic procedures in section 9.2.2.1 or 9.2.2.2 of this appendix. If a quarterly recalibration shows that the value of Yi, the calibration factor at the tested flow rate, differs from the current value of Y by more than 5 percent, repeat the full 3-level calibration of the meter to determine a new value of Y, and apply the new Y value to all subsequent gas volume measurements made with the gas flow meter.
* * * * *10.4 Field Sample Analysis
Analyze the sorbent trap samples following the same procedures that were used for conducting the spike recovery study. The three sections of each sorbent trap must be analyzed separately (i.e., section 1, then section 2, then section 3). Quantify the total mass of Hg for each section based on analytical system response and the calibration curve from section 10.1 of this appendix. Determine the spike recovery from sorbent trap section 3. The spike recovery must be no less than 75 percent and no greater than 125 percent. To report the final Hg mass for each trap, add together the Hg masses collected in trap sections 1 and 2.
* * * * *11.5 [Reserved]
11.6 Calculation of Hg Concentration
Calculate the Hg concentration for each sorbent trap, using the following equation:

Where:
C = Concentration of Hg for the collection period, (μgm/dscm)
M* = Total mass of Hg recovered from sections 1 and 2 of the sorbent trap, (μg)
Vt = Total volume of dry gas metered during the collection period, (dscm). For the purposes of this appendix, standard temperature and pressure are defined as 20 °C and 760 mm Hg, respectively.
11.7 Calculation of Paired Trap Agreeement
Calculate the relative deviation (RD) between the Hg concentrations measured with the paired sorbent traps:

Where:
RD = Relative deviation between the Hg concentrations from traps “a” and “b” (percent)
Ca = Concentration of Hg for the collection period, for sorbent trap “a” (μgm/dscm)
Cb = Concentration of Hg for the collection period, for sorbent trap “b” (μgm/dscm)
* * * * *End Appendix End Supplemental InformationFootnotes
1. On August 22, 2006, EPA proposed to amend Appendix K to allow the data from a pair of sorbent traps to be validated in cases where the third section spike recovery from only one of the traps meets the percent recovery specifications (see 71 FR 49275). EPA proposed to allow the results from the trap that meets the specifications to be used for reporting, provided that a single trap adjustment factor (STAF) of 1.222 is applied. EPA is evaluating the comments received on this proposal and expects to publish the final rule in the summer of 2007.
Back to CitationBILLING CODE 6560-50-C
BILLING CODE 6560-50-C
BILLING CODE 6560-50-C
[FR Doc. 07-4147 Filed 9-6-07; 8:45 am]
BILLING CODE 6560-50-P































Document Information
- Effective Date:
- 11/6/2007
- Published:
- 09/07/2007
- Department:
- Environmental Protection Agency
- Entry Type:
- Rule
- Action:
- Direct final rule.
- Document Number:
- 07-4147
- Dates:
- This rule is effective on November 6, 2007 without further notice, unless EPA receives adverse comment by October 9, 2007. If EPA receives adverse comment, EPA will publish a timely withdrawal in the Federal Register informing the public that some or all of the amendments in this rule will not take effect.
- Pages:
- 51493-51531 (39 pages)
- Docket Numbers:
- EPA-HQ-OAR-2007-0164, FRL-8459-8
- RINs:
- 2060-AO01
- Topics:
- Administrative practice and procedure, Air pollution control, Electric utilities, Environmental protection, Mercury
- PDF File:
- 07-4147.pdf
- CFR: (6)
- 40 CFR 72.2
- 40 CFR 75.15
- 40 CFR 75.20
- 40 CFR 75.57
- 40 CFR 75.81
- More ...